Однородительская дисомия это что
Однородительская дисомия подтверждена для большинства хромосом в кариотипе, хотя клинические аномалии продемонстрированы только для некоторых из них, возможно, отражая позиции одного или нескольких импринтированных генов.
Однородительская дисомия части хромосомы 11 (11р15) связана с развитием синдрома Беквитта-Видемана. Больные дети очень крупные при рождении, имеют увеличенный язык и часто пупочную грыжу.
Выраженная гипогликемия — угрожающее жизни осложнение, так же как развитие злокачественных опухолей почек, надпочечников и печени. Заболевание вызвано избытком отцовского или утратой материнского вклада генов, а также их сочетанием в области хромосомы 11р15, захватывающей локус гена инсули-ноподобного фактора роста II.
Кроме того, описано несколько редких пациентов с муковисцидозом и низким ростом, у которых найдены две идентичные копии материнской хромосомы 7.
В обоих случаях мать случайно оказалась носителем мутации муковисцидоза и, поскольку ребенок получил две материнских копии мутантного гена муковисцидоза и ни одной отцовской копии нормального гена муковисцидоза, у него развивалась болезнь. Задержка роста осталась необъясненной, но могла быть обусловлена потерей неопознанных генов с отцовским импринтингом в хромосоме 7.
Хотя остается неясным, насколько часты однородительские дисомии, они могут объяснить случаи болезни, когда импринтируемая область присутствует в двух копиях от одного родителя.
Таким образом, врачам и генетическим консультантам следует помнить о возможности импринтинга как причине генетического нарушения, особенно в случаях аутосомно-рецессивных заболеваний у пациентов, имеющих только одного подтвержденного родителя — носителя патологической мутации, и в случаях с Х-сцепленными заболеваниями, переданными от отца к сыну или проявившимися в гомозиготной форме у женщин.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Однородительская дисомия
При такой генетической аномалии особь унаследует обе пары хромосом от одного родителя. Описаны различные варианты комбинаций такого наследования, например, передача двух одинаковых или двух разных хромосом. У человека при материнской однородительской дисомии чаще всего передаются 2, 7, 14 и 15 хромосомы, а при отцовской — 6, 11, 15, и 20 хромосомы.
Подобные дефекты могут приводить к серьезной патологии внутриутробного развития, последующему нарушению роста и поведения пациента и специфическим заболеваниям. Существует гипотеза, что однородительская дисомия развивается в результате трисомии плода на ранних сроках беременности. Так как данная генетическая аномалия является высоколетальной, плод сможет выжить, только потеряв лишнюю хромосому. То есть ученые считают, что однородительская дисомия — это механизм выживания плода за счет вытеснения нежизнеспособной трисомичной клеточной линии.
Полученные в результате серьезные нарушения могут затрагивать различные органы и системы. Среди наиболее актуальных заболеваний, которые развиваются вследствие однородительской дисомии, отмечаются муковисцидоз, синдром Прадера-Вилли и синдром Ангельмана.
Муковисцидоз
Главной причиной является мутация в гене CFTR. Однако в литературе описаны случаи развития заболевания в результате однородительской дисомии, в частности, когда оба мутировавших аллеля были унаследованы от одного родителя.
Проявления заболевания зависят от конкретной формы муковисцидоза. Это может быть поражение дыхательного аппарата, кишечника, повышенное отложение соли на коже, симптом барабанных палочек и др. Чтобы узнать, явилось ли заболевание следствием однородительской дисомии или других причин, пациент и его родители проходят генетическое тестирование.
Лечение муковисцидоза включает в себя пожизненную симптоматическую терапию.
Примерно у одной трети всех больных синдромом Прадера-Вилли выявляется однородительская дисомия по 15-й материнской хромосоме. Заболевание дает о себе знать уже в первые годы жизни ребенка. Оно характеризуется задержкой в росте и умственном развитии, нарушением строения половых желез, сниженным мышечным тонусом, ожирением, гинекомастией и др.
Синдром Прадера-Вилли иногда путают с синдромом Дауна, однако для опытного специалиста не составит труда отличить одно заболевание от другого. Чтобы поставить точку в этом вопросе, назначается исследование кариотипа ребенка и родителей, которое может подтвердить или опровергнуть, что болезнь развилась вследствие однородительской дисомии.
Лечение синдрома Прадера-Вилли симптоматическое. Оно включает в себя массаж, работу с логопедом, гормональную терапию и другие методы.
Еще одним частым заболеванием, которое развивается вследствие однородительской дисомии, является синдром Ангельмана. Он проявляется в виде нарушений психического развития, непроизвольными движениями верхних и нижних конечностей, беспричинным смехом. Из-за таких специфичных симптомов синдром Ангельмана еще называют болезнью «счастливой куклы».
Данная патология развивается вследствие однородительской дисомии по 15-й отцовской хромосоме, кроме того, одной из причин её развития ученые называют мутацию в гене UBE3A. Чтобы установить точную причину синдрома Ангельмана, назначается генетическое тестирование.
Эффективного лечения заболевания, как и других видов патологии, связанных с однородительской дисомией, не разработано. Пациентам назначается симптоматическая терапия, которая помогает улучшить качество жизни и позволяет лучше адаптироваться в обществе.
Наследственные пороки развития при однородительских дисомиях
Геномный импринтинг как универсальный механизм функции генов в развитии. Исследование типов и особенностей формирования однородительских дисомий. Изучение генетических аспектов возникновения синдрома Прадера-Вилли. Синдром Энгельмана: пороки развития.
| Рубрика | Биология и естествознание |
| Вид | реферат |
| Язык | русский |
| Дата добавления | 19.05.2015 |
| Размер файла | 23,7 K |

Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
АО «Медицинский университет Астана»
Кафедра молекулярной биологии и медицинской генетики
На тему: «Наследственные пороки развития при однородительских дисомиях»
Выполнила: Уалиева А.Р.
Проверила: Мироедова Э.П.
Однородительские дисомии. Особенности и типы
Механизмы формирования ОРД
Список использованной литературы
Однородительские дисомии. Особенности и типы
Однородительские дисомии были найдены при муковисцидозе и других моногенных заболеваниях, когда оба мутантных аллеля наследовались от одного родителя. В таких случаях дисомия имитирует аутосомно-рецессивное наследование.
Предполагают, что однородительская дисомия является причиной внутриутробной задержки развития, умственной отсталости и микроцефалии. Эти предположения подтверждены молекулярно-биологическими исследованиями.
Существуют два типа ОРД:
К настоящему времени ОРД обнаружены по многим аутосомам и половым хромосомам человека, за исключением хромосом 3, 12, 18 и 19. ОРД по разным хромосомам набора представлена неодинаково. В целом ОРД материнского происхождения встречается в 3 раза чаще, чем отцовского, что отражает более высокую частоту анеуплоидии в ооцитах по сравнению со сперматозоидами Учитывая данные о том, что с возрастом матери увеличивается частота мейотического нерасхождения хромосом, следует предполагать, что этот фактор способствует увеличению риска возникновения ОРД. Поскольку в структуре хромосомных анеуплоидий материнского происхождения преобладают нарушения первого мейотического деления по сравнению со вторым, то можно ожидать преобладание гетеродисомий над изодисомиями. Результаты исследований показывают, что частота гетеродисомий действительно выше. Кроме того, высокий риск формирования ОРД имеют некоторые структурные нарушения хромосом, в частности робертсоновские транслокации. Так, трисомии, обусловленные робертсоновскими транслокациями акроцентрических хромосом, имеют 50%-й риск превращения в ОРД, а однородительские дисомии по хромосоме 13 и другой акроцентрической хромосоме оказываются наиболее частыми. Высокий риск возникновения ОРД имеется также у индивидов с изохромосомами и маркерными хромосомами. Кариотипически «нормальные» клетки, возникающие в результате редукции трисомии или дупликации моносомии, могут приобретать селективное преимущество в росте по сравнению с анеуплоидными клетками, что обеспечивает выживание организмов с таким «спасенным» кариотипом.
Механизмы формирования ОРД
Различают следующие механизмы формирования ОРД:
2. Редукция трисомии. Возникает у первоначально трисомного по какой-либо хромосоме зародыша в результате последующей потери единственной хромосомы, происходящей от одного из родителей, с сохранением двух хромосом другого родителя (рис. 1б). Это событие может происходить как вследствие нерасхождения хроматид в метафазе, так и при отставании хромосомы в анафазе во время первых митотических делений зиготы. Поскольку большинство событий нерасхождения хромосом у человека возникают в I мейотическом делении у матери, то имеется более высокая вероятность возникновения трисомии с двумя разными гомологичными материнскими хромосомами. Следовательно, редукция данной трисомии в результате потери отцовской хромосомы приведет к гетеродисомному типу ОРД материнского происхождения. Следует отметить, что в целом частота ОРД материнского происхождения приблизительно в 3 раза чаще отцовской, а редукция трисомии является наиболее частым механизмом возникновения ОРД у человека.
4. Митотическая рекомбинация (обмен между хроматидами гомологичных хромосом в соматических клетках) или генная конверсия (замена некоторой последовательности ДНК в хромосоме одного родителя гомологичной ей последовательностью ДНК хромосомы другого родителя) являются событиями, которые могут привести к ОРД по отдельным участкам хромосом. В результате рекомбинации между отцовской и материнской хроматидами на ранних стадиях митотически делящейся зиготы и последующей потери другой дочерней клеточной линии может возникнуть сегментная ОРД без мозаицизма. Однако в случае сохранения обеих дочерних I клеток и их последующего размножения может наблюдаться мозаицизм. Особенно это характерно для синдрома Видемана-Беквита, который относительно часто возникает в результате постзиготической рекомбинации гомологичных хромосом в дистальном участке короткого плеча хромосомы 11с образованием сегментной ОРД отцовского происхождения.
Удивительными примерами, иллюстрирующими эти положения, являются хромосомные синдромы Прадера-Вилли и Энгельмана, обусловленные микроделециями в области 15q11-13.
Синдром Прадера—Вилли (СПВ) является наиболее частой причиной генетически обусловленного тяжело протекающего ожирения у детей старше 1 года жизни и у взрослых. Встречается с одинаковой частотой среди обоих полов, регистрируется у лиц разных национальностей и рас. Популяционная частота составляет от 1:10 000 до 1:15 000.
Генетические аспекты возникновения СПВ к настоящему времени считаются достаточно изученными. Установлено 3 варианта нарушения функции участка PWS-AS (сегмент q11—13, ассоциированный с СПВ) в 15-й паре хромосом. В возникновении СПВ всегда «виновата» хромосома отцовского происхождения. В 70% случаев заболевание возникает в результате делеции указанного участка в хромосоме 15 отцовской гаметы. У 20% больных СПВ развивается в результате полного отсутствия в клетках ребенка отцовской хромосомы 15 и замены ее дуплицированной (копированной) материнской хромосомой. Подобный феномен называется материнской дисомией. В 5% случаев ответственной за возникновение патологии является функциональная дезактивация у плода структурно нормального участка PWS-AS отцовской хромосомы. Это происходит в результате его метилирования, т. е. присоединение к цитозиновым основаниям групп-CH.
К этим ранним проявлениям синдрома относятся:
— вялое шевеление плода,
— слабость сосательного рефлекса вскоре после рождения,
— развитие мышечной гипотонии в первые месяцы жизни.
В связи с нарушением акта сосания при отсутствии зондового питания у ребенка могут быстро развиться дистрофия и другие дефицитные состояния. В связи с мышечной гипотонией страдает моторное развитие.
Уже на 1-м году жизни могут выявляться различные дизморфии лица и конечностей, гипогонадизм и/или гипогенитализм. Наличие у ребенка первых месяцев жизни указанных симптомов нередко приводит врача к ошибочной диагностике пре- и перинатального поражения ЦНС, инфекционного процесса, так как часты нарушения терморегуляции; различных вариантов наследственных миопатий, врожденного гипотиреоза и других болезней.
В большинстве случаев клиническая диагностика СПВ осуществляется после годовалого возраста ребенка, когда развивается 2-я фаза заболевания, характеризующаяся появлением у больного постоянного чувства голода. У ребенка постепенно и стойко формируется поведение «постоянного поиска пищи».
К крупным порокам относятся следующие:
* характерные лицевые симптомы (долихоцефалия с уменьшением битемпорального диаметра, миндалевидный разрез глаз, небольшой рот с опущенными вниз углами и тонкой верхней губой, страбизм);
* задержка нервно-психического развития до 6 лет, умеренное снижение интеллекта и проблемы обучения в школьном возрасте;
* проблемы при кормлении в первые месяцы жизни с последующей нормализацией сосания в течение грудного периода;
* изменения со стороны половой сферы (крипторхизм, гипоплазия мошонки или маленькие тестикулы у мальчиков; гипоплазия малых губ и клитора у девочек; нарушения полового созревания);
* мышечная гипотония центрального генеза в раннем детстве;
* прогрессирующее ожирение в возрасте от 1 года до 6 лет.
К малым порокам относятся следующие:
* снижение двигательной активности плода и инфантильная летаргия;
* снижение пигментации кожи и волос в сравнении с родителями;
* следы «потертостей» кожных покровов (;
* поперечная ладонная складка;
* низкорослость к 15 годам с учетом длины тела других членов семьи;
* нарушения сна и апноэ во время сна;
* маленькие стопы и/или кисти;
* дефекты артикуляции и речи
* густая, вязкая слюна;
геномный импринтинг однородительский дисомия
Синдром Энгельмана возникает вследствие потери нормальных материнских копий генов в определенной области 15 хромосомы. Чаще всего это происходит путем делеции сегмента этой хромосомы. Другими причинами возникновения заболевания могут быть одноотцовская дисомия, транслокация или мутация одного гена в этой области.
Наиболее распространенным генетическим дефектом, который приводит к появлению синдрома Энгельмана, является: делеция 4Mb (мега) материнской копии хромосомной области 15q11-13, которая вызывает отсутствие экспрессии родительской копии UBE3A в мозге.
Характерны для всех (100%):
— тяжелая функциональная задержка развития;
— нарушение речевых функций (полное отсутствие или минимальное использование языка); невербальные навыки развиты лучше, чем вербальные;
— расстройство моторных функций (нарушения движения, равновесия), как правило, атаксия ходы и (или) дрожание конечностей;
— поведенческие отклонения: сочетание частого беспричинного смеха (улыбки), счастливого состояния, легкой возбудимости, частого хлопанья в ладоши, гипермоторного поведения, низкий уровень концентрации внимания.
Частые черты (характерны более чем для 80% больных):
— задержка, непропорциональное увеличение окружности головы, которое, как правило, приводит к возникновению микроцефалии (абсолютной или относительной) до возраста 2 лет;
— приступы судорог, которые обычно появляются в возрасте менее трех лет;
— аномальные результаты ЭЭГ (электроэнцефалограммы), характерной особенностью которых является большая амплитуда и низкая временная динамика волн.
— гипопигментация кожи и глаз;
— нарушение контроля над движениями языка, трудности при сосании и глотании;
— гиперактивные сухожильные рефлексы;
— проблемы с питанием в раннем детстве;
— подняты, согнуты во время шествия руки;
— выдвинута нижняя челюсть;
— повышенная чувствительность к теплу;
— широкий рот, широкий интервал между зубами;
— частые слюнотечения, высунутый язык;
— постоянное желание пить;
— усиленные жевательные движения;
Необходимость изучения причин эмбриональной гибели у человека продиктована довольно высоким уровнем репродуктивных потерь. При этом цитогенетическое обследование в среднем у 50% погибших эмбрионов выявляет аномалии кариотипа, однако причина гибели другой половины, как правило, остается неясной.
Суть рабочей гипотезы, проверке которой было посвящено глобальное исследование, состояла в том, что у части цитогенетически нормальных погибших зародышей человека кариотип может быть псевдонормальным, то есть нести внешне никак не детектируемую ОРД по какой-либо хромосоме набора. Вследствие явления геномного импринтинга, достаточно хорошо установленного у человека только по некоторым хромосомам набора (особенно по хромосомам 7, 11, 14 и 15), не исключена возможность того, что ОРД по другим, еще не идентифицированным до сих пор хромосомам, может быть причиной ранней эмбриональной гибели зародышей.
Список использованной литературы
1. Назаренко С.А., Никитина Т.В. Униродительская дисомия в исследовании проблемы геномного импринтинга у человека. В сб. Генетика человека и патология,- Томск. 1997- С.36-48.
2. Перепечина И.О., Гришечкин С.А. Вероятностные расчеты в ДНК-дактилоскопии: Методические рекомендации. М.: ЭКЦ МВД России, 1996,-16с.
3. Редина O.E. Геномный импринтинг как универсальный механизм функции генов в развитии. // Генетика.- 1996,- Т.32,- N 5,- С.609-613.
4. Сапиенца К. Геномный импринтинг // В мире науки,- 1990,- №12,-С. 14-20.
5. В.Н.Горбунова Медицинская-генетика
Размещено на Allbest.ru
Подобные документы
Цитогенетические заболевания, вызванные микроделецией и дисомией хромосом. Клинические проявления синдромов Прадера-Вилли, Ангельмана, Ди Джорджи. Геномный импринтинг, наследственные заболевания; медицинская помощь, риски дальнейших проявлений патологий.
курсовая работа [6,3 M], добавлен 07.06.2011
Исследование современных методов и проблем диагностики наследственной патологии: наследственные заболевания и болезни импринтинга. Изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
курсовая работа [1,3 M], добавлен 02.06.2011
Описания специфичного комплекса уродств и пороков развития у детей, родившихся от родителей, злоупотребляющих спиртными напитками. Исследование тяжелых нарушений психического развития. Характеристика особенностей возникновения алкогольного синдрома плода.
презентация [1,6 M], добавлен 02.10.2014
Тератогенные факторы, вызывающие пороки эмбрионального развития человека. Аномалии в строении организма. Никотин, алкоголь и наркомания и их воздействие на организм. Ранняя детская смертность. Взаимосвязь между тератогенами и здоровьем человека.
научная работа [40,8 K], добавлен 04.07.2009
Эволюция представлений о гене. Основные методы идентификации генов растений. Позиционное клонирование (выделение) генов, маркированных мутациями. Выделение генов, маркированных делециями методом геномного вычитания и с помощью метода Delet-a-gen.
контрольная работа [937,4 K], добавлен 25.03.2016
Однородительская дисомия, что это: примеры
Однородительская дисомия это
При мозаичных трисомиях по любой из хромосом, а также в половине случаев моносомии Х, независимо от типа мозаицизма и доли анеуплоидных клеток, вторая клеточная линия всегда является диплоидной. Как уже отмечалось, мейотический плацентарный мозаицизм типа 3 сопровождается восстановлением диплоидного кариотипа у плода вследствие утраты хромосом из триады.
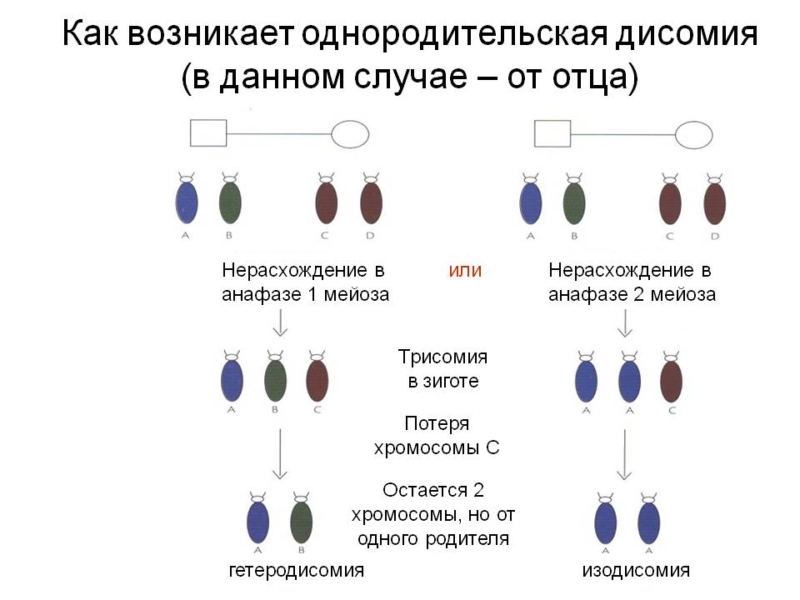
Коррекция трисомии в 1/3 случаев может приводить к возникновению однородительской дисомии — ОРД (uniparental disomy, или upd), т. е. наличию в диплоидном хромосомном наборе двух гомологичных хромосом только от одного из родителей. Возможные механизмы образования ОРД представлены на рисунке 7.8.
Следует отметить, что ОРД является результатом, по меньшей мере, двух последовательных независимых событий аномальных сегрегаций хромосом, происходящих в гаметогенезе и во время постзиготических митотических делений. Аналогично возникает ОРД и в случаях аберраций хромосом, таких как изохромосомы или Робертсоновские транслокации.
ОРД подразделяют на гетеро- и изодисомию. При гетеродисомии (нерасхождением хромосом в первом делении мейоза) (рис. 7.7, а; 7.8), в диплоидном наборе представлены оба гомолога от одного родителя. Изодисомия, т. е. удвоение одного из родительских гомологов, возникает вследствие нерасхождения хроматид во втором делении мей- оза или дупликацией хромосомы при дроблении моносомной зиготы (рис. 7.7, б; 7.8).

Рис. 7.8. Механизмы формирования однородительской дисомии
Крайним вариантом ОРД является однородительская диплоидия, т. е. утрата целого хромосомного набора отцовского или материнского происхождения. Андрогения (отсутствие материнского и удвоенный отцовский хромосомный набор) возникает в результате оплодотворения безъядерной яйцеклетки диплоидным спермием или дупликации генома гаплоидного спермия после оплодотворения.
При андрогении собственно эмбрион и амнион не формируются, однако отмечается активное разрастание ворсинок хориона, гиперплазия трофобласта c отеком стромы и отсутствием капилляров. При дальнейшем развитии такого продукта зачатия, получившего название «полный пузырный занос», его клетки зачастую озлока- чествляются, давая начало быстро метастазирующей опухоли — хорионэпителиоме.
Гиногения (удвоен материнский, отсутствует отцовский набор хромосом) — продукт партеногенетического развития овулировавшей неоплодотворенной яйцеклетки. Гиногения у человека ведет к дезорганизации плодного яйца, хаотичному росту эмбриональных тканей, образованию тератомы при недоразвитии или полном отсутствии оболочек плода.
Таким образом, аномальная сегрегация хромосом при дроблении зиготы и на более поздних стадиях эмбриогенеза приводит к моза- ицизму. При этом мозаицизм независимо от происхождения может быть ограниченным (в одной ткани) или истинным (во всех тканях).
Для объяснения механизмов возникновения мозаичных эмбрионов и мозаицизма, ограниченного плацентой, предложено несколько гипотез. Однако до сих пор не установлены факторы, способствующие нерасхождению хромосом при дроблении зиготы, а также не определен вклад каждого из возможных механизмов в формирование мозаичных особей, не выяснены особенности их эмбрионального развития.
Болезни с нетрадиционным типом наследования
В последние годы стало очевидным, что далеко не все случаи наследственной патологии у человека можно рассматривать как результат менделирующих генных мутаций, хромосомных аномалий или как мультифакториальные заболевания (МФЗ).
В настоящее время описано достаточно много заболеваний, которые в современной классификации наследственной патологии человека объединяют в отдельную группу: болезни с нетрадиционным типом наследования.
Среди них различают: болезни импринтинга, митохондриальные болезни, болезни экспансии тринуклеотидных повторов с явлением антиципации и др.
Болезни импринтинга
Особенности наследования и фенотипического проявления при болезнях импринтинга обусловлены явлением геномного импринтинга (ГИ) (импринтинг от англ. imprinting — запечатление).
Явление геномного импринтинга связывают со специфическими изменениями хромосом или их участков во время образования мужских и женских гамет.
Этим объясняется дифференциальная маркировка отцовских и материнских хромосом у потомков.
Точные механизмы дифференциальной маркировки хромосом или их участков в сперматогенезе или овогенезе пока окончательно не выяснены. Однако, немаловажная роль, вероятно, принадлежит процессам специфического метилирования цитозиновых оснований ДНК, выключающим транскрипцию гена.
Импринтированные участки в хромосомах определенного родительского происхождения (отцовских иди материнских) избирательно репрессируются у потомка.
В связи с этим фенотипически проявляется только информация, полученная от другого родителя, т.е. имеет место моноаллельная экспрессия. Следовательно, фенотипическое проявление мутантного аллеля зависит от того с какой половой клеткой (яйцеклеткой или сперматозоидом) он был передан потомку.
Явлением ГИ объясняется, например, избирательная инактивация у млекопитающих отцовской Х-хромосомы в клетках провизорных органов (см. гл. 7.5.4.). В клетках самого зародыша имеет место равновероятная инактивация отцовской и материнской Х-хромосом (см. рис. 3.78).
Таким образом, следствием ГИ (дифференциальной маркировки в гаметогенезе родителей и последующей избирательной инактивации у потомков участков хромосом) является функциональная неравноценность в генотипе потомка аллелей разного родительского происхождения.
Связь этиологии ряда наследственных заболеваний с феноменом ГИ может быть прослежена на разных уровнях организации генетического материала.
На геномном уровне организации наследственного материала доказательством роли ГИ в патологии служит различное фенотипическое проявление триплоидных состояний при разном соотношений гаплоидных наборов отцовского и материнского происхождения.
У диандрических триплоидов (соотношение числа гаплоидных наборов отца и матери 2:1) и у дигенических триплоидов (соотношение 1:2) патологические отклонения в развитии плаценты и собственно зародышевых тканей проявляются по-разному.
Это свидетельствует о неравноценности функционирования гаплоидных наборов отца и матери в тканях зародыша и плаценты (см, разд.7.5.4 и 7.6.1).
Связь феномена ГИ с патологией на уровне отдельных хромосом можно проследить в случае однородительской дисомии (ОРД), при которой происходит удвоение хромосомы одного из родителей при утрате гомологичной хромосомы другого родителя.
В основе возникновения ОРД лежит нарушение процессов гаметогенеза. При нерасхождении сестринских хроматид в анафазе II мейоза появляются гаметы, в галлоидном наборе которых присутствуют две генетически идентичные хромосомы (изодисомия).
В случае нерасхождения гомологичных хромосом в анафазе I мейоза образуются гаметы, в гаплоидном наборе которых имеется пара гомологичных, генетически неидентичных хромосом (гетеродисомия).
В обоих случаях гаметы данного индивида дисомны по одной из хромосом.
При оплодотворении дисомных гамет нулисомными по той же хромосоме подовыми клетками происходит комплемеитация гамет, приводящая к возникновению нормального диплоидного кариотипа зиготы. Однако в генотипе такой зиготы присутствует двойной набор генов данной хромосомы, происходящих от одного, а не от обоих родителей.
Иногда оплодотворение дисомных гамет нормальными половыми клетками сопровождается «коррекцией трисомии» в результате потери сверхчисленной хромосомы.
Если при этом сохраняются две хромосомы, пришедшие от одного родителя, то наблюдается явление ОРД.
Наконец, состояние ОРД по отдельным локусам хромосом может возникать в результате соматической рекомбинации — кроссинговера между хроматидами гомологичных хромосом, происходящего в соматических клетках (см. рис 3.73).
Когда хромосома не содержит импринтированных участков, при ОРД по данной хромосоме может не наблюдаться аномалий фенотипа.
Исключением может быть проявление аутосомно-рецессивного заболевания как результат гомозиготизации по рецессивному аллелю при изодисомии.
Если хромосома содержит импринтированные участки, то при возникновении однородительской дисомии локализованные в них аллели могут быть либо экспрессированы, либо инактивированы в зависимости от родительского происхождения ОРД. Это может стать причиной возникновения патологических отклонений в развитии организма. Фенотипическое проявление при ОРДмат и ОРДотц может быть сходным или прямо противоположным.
Возможен летальный эффект уже на ранних сроках развития.
В настоящее время эффект импринтинга установлен достаточно определенно для четырех хромосом человека 15, 11, 7, 14.
Так в проксимальном отделе длинного плеча 15-й хромосомы имеется район, подверженный импринтингу. Мутации, связанные с микроделециями в этом районе, приводят к развитию у человека синдрома Прадера — Вилли, при котором у пациентов наблюдается умственная отсталость, мышечная гипотония, сильное ожирение, гипогонадизм, низкий рост, акромикрия (непропорционально малые размеры дистальных отделов конечностей).
В настоящее время описано более 30 случаев синдрома Прадера —Вилли, когда у пациентов определяется ОРДмат 15. Считается, что ОРДмат 15 является причиной 20—25% всех случаев этого синдрома. Большая же часть остальных случаев заболевания связана с делецией сегмента 15qll — ql3 отцовской хромосомы.
Указанный пример свидетельствует об активной экспрессии соответствующего участка 15-й хромосомы исключительно отцовского происхождения. В материнской же хромосоме он метилирован и репрессирован.
Делеция другого участка, также расположенного в сегменте 15qll — ql3, но в 15-й хромосоме материнского происхождения, в 70% случаев приводит к развитию синдрома Энгельмана (синдрома «счастливой куклы»), характеризующегося глубокой умственной отсталостью с резкими судорожными движениями и неадекватной счастливой улыбкой.
В 2% случаев этот синдром обусловлен ОРДотц15.
Из сказанного выше следует, что в проксимальном районе длинного плеча 15-й хромосомы имеются близкорасположенные и противоположно импринтированные локусы, отвечающие за возникновение фенотипически различных синдромов Прадера — Вилли и Энгельмана.
Таким образом импринтироваться могут участки хромосом разного родительского происхождения, что и определяет нетрадиционное наследование многих патологических состояний, обусловленных мутациями локусов, подверженных импринтингу.
Митохондриальные болезни
Начиная с конца 80-х годов XX века получены убедительные доказательства связи некоторых видов наследственной патологии у человека с мутациями митохондриальной ДНК (см. гл. 4.1) В зависимости от типа мутаций митохондриальные болезни разделяют на 4 группы:
а) болезни, вызванные точковыми мутациями, приводящими к замене консервативных аминокислот в собственных белках митохондрий. К ним относятся пигментный ретинит и нейроофтальмопатия Лебера, при которой наступает двусторонняя потеря зрения. Выраженность клинических признаков у больных этими заболеваниями коррелирует с количеством мутантной мтДНК, которое у разных больных может варьировать от 5 до 100% всей мтДНК;
б) болезни, вызванные мутациями в генах т-РНК, приводящими к многочисленным дегенеративным заболеваниям с различной степенью тяжести клинических проявлений, коррелирующей с количеством мутантной мтДНК;
в) болезни вызванные делениями и дупликациями участков митохондриалъных генов. У человека описано тяжелое заболевание молодого и среднего возраста — отсроченная кардиопатия, при которой обнаружены делеции мтДНК кардиоцитов.
Заболевание носит семейный характер.
В ряде случаев предполагается Х-сцепленное наследование, что позволяет думать о существовании ядерного гена, мутация которого вызывает делению до 50% мтДНК кардиоцитов;
г) болезни, вызванные снижением числа копий мтДНК, что является следствием определенных мутаций. К данной группе относятся летальная инфантильная дыхательная недостаточность и синдром молочнокислого ацидоза, при которых число копий мтДНК снижается до 1—2% от нормы.
Снижение содержания мтДНК в клетках различных органов приводит к развитию миопатий, нефропатий, печеночной недостаточности и т.д. вследствие ослабления синтеза белков, кодируемых мтДНК.
Изменения в ДНК митохондрий сопровождаются нарушением их функций, связанных с клеточным дыханием. Это определяет характер и степень тяжести клинических проявлений митохондриалъных болезней.
Выдвинута также гипотеза о том, что накопление спонтанно возникающих мутаций мтДНК является звеном механизмов старения и развития дегенеративных процессов у человека.
Болезни экспансии тринуклеотвдных повторов с явлением антиципации
Под генетической антиципацией (или упреждением) понимается более раннее проявление и возрастание тяжести симптомов наследственного заболевания в последующих поколениях родословной.
Антиципация реально проявляется при определенных видах моногенной неврологической патологии, а также при некоторых мультифакториальных заболеваниях.
В начале 90-х годов XX века при исследовании ряда тяжелых неврологических заболеваний были обнаружены «динамические» мутации с экспансией (резким увеличением числа копий) тринуклеотидных повторов у индивидов в последующих поколениях родословной. Развивающиеся в результате таких мутаций наследственные заболевания характеризуются четко выраженным проявлением антиципации.
Феномен экспансии числа тринуклеотидных повторов был впервые обнаружен при исследовании синдрома Мартина—Белла или синдрома фрагильной (ломкой) Х-хромосомы, основным фенотипическим проявлением которого является умственная отсталость.
Синдром ломкой Х-хромосомы характеризуется довольно широкой распространенностью в популяции (1:1000) и необычным характером наследования. Лишь у 80% мужчин-носителей мутантного локуса имеются клинические и цитогенетические признаки заболевания. 20% носителей как клинически, так и цитогенетически нормальны, но после передачи мутации всем своим дочерям они могут иметь пораженных внуков. Неэкспрессируемый мутантный ген в таком случае становится экс-прессируемым в последующих поколениях.
Таким образом мутантный ген при синдроме ломкой Х-хромосомы может существовать в двух формах, отличающихся по своей пенетрантности.
Одна — фенотипически не проявляющаяся — премутация, которая при прохождении через женский мейоз превращается в другую форму — полную мутацию. При таком необычном способе наследования и фенотипического проявления мутантного гена, отличном от классического Х-сцепленного наследования, обнаруживается феномен антиципации — более тяжелое проявление заболевания в последующих поколениях.
В основе клинических проявлений и цитологической нестабильности в локусе, ответственном за синдром ломкой Х-хромосомы, лежит многократное увеличение повторов тринуклеотида ЦГГ.
В норме число повторов колеблется от 5 до 50. Премутация — неэкспрессируемая форма — характеризуется увеличением числа повторов до 50—200. Возрастание числа повторов тринуклеотида ЦГГ свыше 200 приводит к клинической манифестации заболевания и цитогенетическому проявлению ломкой Х-хромосомы.
Как правило, у пораженных лиц наблюдается также аномальное метилирование ДНК, приводящее к репрессированию гена.
Интересно, что переход от состояния премутации к полной мутации возникает при передаче от матери, причем экспансия ЦГГ-повторов значительно выше при передаче от матери к сыну, чем от матери к дочери.
Антиципация, характерная для синдрома ломкой Х-хромосомы, объясняется четкой связью между числом тринуклеотидных повторов и тяжестью клинических проявлений заболевания с цитологической экспрессией ломкости Х-хромосомы.
Митохондриальное наследование
Важными особенностями митохондриального типа наследования патологии являются:
–наличие патологии у всех детей больной матери;
– рождение здоровых детей у больного отца и здоровой матери. Указанные особенности объясняются тем, что митохондрии наследуются только от матери.
Доля отцовского митохондриального генома в зиготе составляет ДНК от 0 до 4 митохондрий, а материнского генома — ДНК примерно от 2500 митохондрий.
К тому же, после оплодотворения репликация отцовской ДНК блокируется.
В настоящее время геном митохондрий секвенирован. Он содержит 16 569 пар оснований и кодирует две рибосомные РНК (12S и 16S), 22 транспортные РНК и 13 полипептидов – субъединиц ферментативных комплексов окислительного фосфорилирования. Другие 66 субъединиц дыхательной цепи кодируются в ядре.
Примеры заболеваний с митохондриальным типом наследования (митохондриальные болезни): атрофия зрительного нерва Лебера, синдромы Лея (митохондриальная миоэнцефалопатия), MERRF (миоклоническая эпилепсия), кардиомиопатия дилатационная семейная.
Родословная пациента с митохондриальным типом наследования патологии (атрофия зрительного нерва Лебера) в четырёх поколениях представлена на рис. 4–13.
Рис 04 13 Родословная с митохондриальным типом наследования заболевания»

Рис.4–13.Родословная с митохондриальным типом наследования заболевания.
Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной.
Наследственные пороки развития при однородительских дисомиях
Примеры моногенных заболеваний, наиболе часто встречающихся в клинической практике.
Фенилкетонурия
Все формы фенилкетонурии являются результатом недостаточности ряда ферментов. Их гены транскрибируются в гепатоцитах и наследуются по аутосомно‑рецессивному типу.
Наиболее частая форма фенилкетонурии возникает при мутациях гена фенилаланин 4‑монооксигеназы (фенилаланин 4-гидроксилаза, фенилаланиназа). Самый распространённый тип мутаций – однонуклеотидные замены (миссенс‑, нонсенс‑мутации и мутации в сайтах сплайсинга).
Ведущее патогенетическое звено фенилкетонурии – гиперфенилаланинемия с накоплением в тканях токсических продуктов метаболизма (фенилпировиноградной, фенилуксусной, фенилмолочной и других кетокислот). Это ведёт к поражению ЦНС, нарушению функции печени, обмена белков, липо- и гликопротеинов, метаболизма гормонов.
Проявляется фенилкетонурия: повышенной возбудимостью и гипертонусом мышц, гиперрефлексией и судорогами, признаками аллергического дерматита, гипопигментацией кожи, волос, радужки; «мышиным» запахом мочи и пота, задержкой психомоторного развития.
У нелеченых детей формируется микроцефалия и умственная отсталость. С этим связано другое название заболевания – фенилпируватная олигофрения.
Лечение фенилкетонурии проводится с помощью диетотерапии (исключением или снижением содержания в пище фенилаланина). Диету необходимо соблюдать с момента установления диагноза (первые сутки после рождения) и контролировать содержание фенилаланина в крови не менее 8–10 лет.Гемофилия А(см.статью «Гемофилия» в приложении «Справочник терминов»)
Синдром Марфана
Частота синдрома Марфана находится в диапазоне 1:10 000–15 000. Наследуется синдром по аутосомно‑доминантному типу. Причина синдрома – мутация гена фибриллина (FBN1). Идентифицировано около 70 мутаций этого гена (преимущественно миссенс‑типа).
Мутации различных экзонов гена FBN1 вызывают разные изменения фенотипа, от умеренно выраженных (субклинических) до тяжёлых.
Проявляется синдром Марфана генерализованным поражением соединительной ткани (поскольку фибриллин широко представлен в матриксе соединительной ткани кожи, лёгких, сосудов, почек, мышц, хрящей, сухожилий, связок); поражением скелета, высоким ростом, диспропорционально длинными конечностями, арахнодактилией, поражениями сердечно‑сосудистой системы, расслаивающимися аневризмами аорты, пролапсом митрального клапана, поражением глаз: вывихами или подвывихами хрусталика, дрожанием радужки.
Гемоглобинопатия S
Гемоглобинопатия S (аутосомно‑рецессивное наследование) распространена в странах так называемого малярийного пояса Земли.
Это объясняется тем, что гетерозиготы по HbS резистентны к тропической малярии. В частности, носители HbS распространены в Закавказье и Средней Азии, в России максимальная частота гетерозиготных носителей HbS отмечена в Дагестане.
Причиной HbS является замещение одного основания в 6‑м триплете (миссенс‑мутация) ‑цепи глобина. Это приводит к замене глутаминовой кислоты на валин.
Такой Hb имеет крайне низкую растворимость. Внутриклеточно из HbS образуются кристаллические тактоиды. Они и придают эритроцитам форму серпа. Отсюда название болезни – «серповидно-клеточная анемия».
Гетерозиготные носители HbS в обычных условиях здоровы, но при пониженном pO2 (кессонные работы, условия высокогорья и т.д.) или при гипоксемии (ВПР сердца, дыхательная недостаточность, длительный наркоз и т.п.) развивается гемолитическая анемия.
Гомозиготы страдают тяжёлой гемолитической анемией с 4–6‑месячного возраста.
В результате тромбоза капилляров или венул серповидными эритроцитами развиваются трофические язвы (часто на голени), боли в животе, поражение сердца, глаз. Характерны поражения костно‑суставной системы, гепатоспленомегалия.
Муковисцидоз
Муковисцидоз — множественное поражение экзокринных желёз, сопровождающееся накоплением и выделением ими вязких секретов.
Среди новорождённых частота муковисцедоза составляет 1:1500–1:2000. Кистозный фиброз является одним из самых распространённых моногенных заболеваний в Европе. Наследуется муковисцидоз по аутосомно‑рецессивному типу. Известно более 130 мутантных аллелей; наиболее частая мутация – delF508.
Она приводит к отсутствию фенилаланина в 508-м положении трансмембранного регуляторного белка. В зависимости от типа мутаций и их локализации функция гена может быть полностью или частично нарушена. При этом расстраивается регуляция переноса Cl– через мембраны эпителиальных клеток (транспорт Cl– тормозится, а Na+ усиливается).
Болезнь характеризуется закрытием протоков желёз вязким секретом, который образуется в связи с повышенной резорбцией Na+ клетками протоков экзокринных желёз.
Нередко в протоках образуются кисты и развивается воспаление. При хроническом течении в железах развивается избыток соединительной ткани (склероз). У новорождённых нередко выявляется непроходимость кишечника (мекониальный илеус). У детей наиболее часто развивается лёгочная или лёгочно‑кишечная форма заболевания.
Оно проявляются повторными бронхитами, пневмониями, эмфиземой лёгких, а также нарушениями полостного и пристеночного пищеварения, вплоть до развития синдрома мальабсорбции (синдром нарушенного всасывания). При длительном течении развиваются дыхательная недостаточность, цирроз печени, портальная гипертензия, нередко приводящие к смерти.
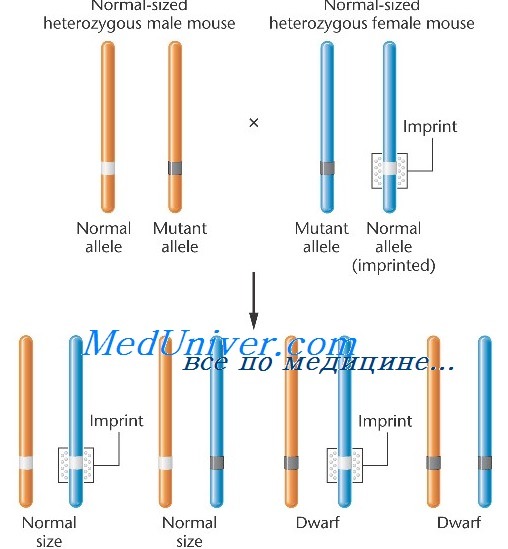
Однородительская дисомия (UPD)
В период классических генетических исследований некоторые мыши- мутанты с хромосомными транслокациями, первоначально использовавшиеся для картирования генов на хромосомах, демонстрировали специфичный в отношении родителя фенотип, когда определенные хромосомные участки были унаследованы как дупликации хромосомы одного родителя в отсутствие хромосомы другого родителя, что получило определение однородительской дисомии (uniparental disomy, UPD) рис.
Возможный механизм дисомии — элиминация лишней хромосомы у плода с трисомией на ранних стадиях эмбриогенеза.
Болезнь проявляется в том случае, если элиминируется лишняя хромосома, происходящая из нормальной гаметы.
Отцовская 15-я хромосома у таких больных отсутствует.
У пациентов, наследующих обе гомологичные хромосомы (или их сегменты) от одного и того же родителя (однородительская дисомия — uniparental disomy, UPD), наблюдается потеря экспрессии некоторых генов, которые экспрессируются только в материнских аллелях (в случае отцовской UPD), и повышение уровня экспрессии отцовских генов.
Изучение необычных пациентов не только установило случаи UPD для дополнительных хромосом, но и привело в 1989 г.к предположению, что UPD вызывает заболевания благодаря изменениям в эпигенотипе и нарушению геномного импринтинга ( Nicholls et al., 1989 ).


