Нф1 что это диагноз
При нейродерматозах повреждения головного мозга сопровождаются поражением кожи. К этой группе заболеваний относят нейрофиброматоз (заболеваемость составляет 1: 5 000) и туберозный склероз (заболеваемость составляет 1:10 000). А. Нейрофиброматоз 1 типа (НФ 1)
1. Определение. НФ 1 (болезнь Реклингхаузена) — аутосомно-доминантное заболевание с генной локализацией в 17 хромосоме. Около 50 % случаев обусловлены новыми мутациями. Согласно критериям Национального института здоровья, диагноз НФ 1 может быть установлен при наличии двух или более из семи перечисленных ниже признаков: пятна цвета «кофе с молоком», нейрофибромы, веснушки в подмышечной области, характерные костные аномалии, глиома зрительного нерва, узелки на радужной оболочке, установленный диагноз НФ 1 у ближайшего родственника.
2. Обследование
Анамнез. В половине случаев имеет место семейный анамнез НФ 1, причем большое значение может иметь расспрос о родимых пятнах и опухолях. Многие дети, страдающие НФ 1, несколько отстают в развитии и имеют проблемы с учебой. Часто больные жалуются на запоры и головную боль. Снижение остроты зрения указывает на глиому зрительного нерва.
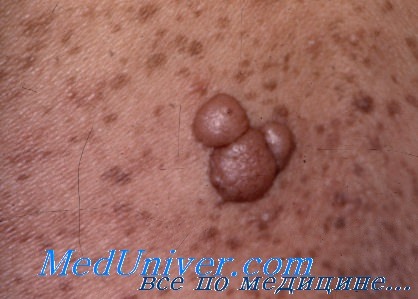
Объективное обследование. Одиночные изолированные пятна цвета «кофе с молоком» часто встречаются у здоровых людей. У детей с НФ 1 определяют не менее 6 пятен с наименьшим диаметром 5 мм, которые наиболее часто располагаются на туловище. Подмышечные и паховые области при НФ 1 покрыты веснушками (пигментными пятнышками). Сетчатые нейрофибромы соединяются опухолевыми тяжами, что может стать причиной деформации заинтересованных структур. Дермальные нейрофибромы, кожа над которыми имеет фиолетовую или красную окраску, представляют собой опухоли мягкой консистенции.
У 2 % пациентов с НФ 1 имеет место кифосколиоз, а в 0,5—1 % случаев заболевания отмечаются ложные суставы. Гипертензия может развиться вследствие стеноза почечной артерии или феохромоцитомы. С целью исключения опухолей ЦНС необходимо повторное неврологическое обследование. Осмотр офтальмолога в свете щелевой лампы необходим для обнаружения гамартром радужной оболочки (узелки Лиша).
Лабораторные исследования. Бели в семье имеются случаи нейрофиброматоза, с целью ранней диагностики может быть выполнен хромосомный анализ. При любых проявлениях дисфункции ЦНС или изменении остроты зрения, свидетельствующей о глиоме зрительного нерва, показана МРТ. При МРТ головного мозга у детей с НФ 1 частой находкой являются зоны усиленного сигнала, расположенные в базальных ядрах. При наличии признаков задержки развития или снижения академической успеваемости показано психологическое обследование.
3. Течение заболевания. У грудного ребенка с НФ 1 могут обнаруживаться пятна цвета «кофе с молоком», плексиформная нейрофиброма, глаукома. В младшем детском возрасте становится заметным увеличение размера головы, отставание в росте, выявляются ложные суставы. Узелки радужной оболочки, веснушки, головные боли, запоры и трудности в учебе характерны для раннего школьного возраста. Нейрофибромы, сколиоз и гипертония проявляются у подростков.
4. Дифференциальный диагноз. Вариантом нормы следует считать наличие единичных (до четырех) пятен цвета «кофе с молоком». Отдельные нейрофибромы и глиомы зрительного нерва могут наблюдаться у детей без каких-либо других проявлений НФ 1. Если в младенческом возрасте на теле у ребенка обнаруживают более пяти пятен характерной окраски, то вероятность развития других признаков НФ 1 составляет 50-60 %.
Нейрофиброматоз 2 типа (НФ 2) — наследуемое по аутосомно-доминантному типу заболевание с генной локализацией в 22 хромосоме. Встречается намного реже, чем НФ 1. Первичным проявлением заболевания является шваннома вестибулярной порции восьмого черепного нерва, а также менингиомы и глиомы ЦНС. Изменения кожи в виде пятен цвета «кофе с молоком» при НФ 2 встречаются реже, чем при НФ1. Симптомы заболевания развиваются чаще в подростковом и юношеском возрасте.
Нейрофиброматоз 1 типа
Нейрофиброматоз 1 типа (НФ1, болезнь Реклингхаузена, периферический НФ) – генетическое заболевание, которое встречается примерно у 1 из 2500-3500 новорожденных детей. Это самое распространенное генетическое заболевание среди редких. По данным американской ассоциации CTF каждый день рождается около 120 человек с нейрофиброматозом 1 типа. Это значит, что новый пациент с НФ1 рождается каждые 12 минут.
Диагноз нейрофиброматоз 1 типа может быть поставлен при наличии сочетания ДВУХ И БОЛЕЕ симптомов:

Данная информация носит ознакомительный характер и не может быть использована для самодиагностики, диагноз может поставить только врач!
Указанные признаки могут встречаться в любом сочетании, но ни один из них в отдельности не является достаточным для диагностики НФ1. Также отсутствие какого-либо из признаков (например, пятен цвета «кофе с молоком», которые присутствуют примерно у 99% больных НФ1) не препятствует постановке диагноза.
Также при нейрофиброматозе 1 типа у пациентов встречаются: эпилептические приступы, опухоли центральной нервной системы (ЦНС), вишневые ангиомы (пятна Кэмпбелла де Моргана), ксантогранулемы, низкорослость, раннее половое созревание и другие более редкие проявления, не являющиеся диагностическими, но требующими внимания.
Симптомы НФ1 проявляются у пациентов индивидуально в различные периоды жизни и практически на всем ее протяжении – часть из них формируется в течение первых двух лет жизни, другие – к 5-7 годам, и так далее. Например, симптомы, имеющие кожные проявления, чаще всего проявляются при рождении, в младенческом возрасте или до достижения ребенком возраста 10 лет. Появление пигментных пятен, пятен кофейного цвета и узелков Лиша, как правило, не представляют опасности для здоровья, хотя могут стать причиной психологического дискомфорта. Примерно в 10-15 лет характерным становится появление нейрофибром, образование которых может проходить внутри тела и затрагивать деятельность различных органов. Гормональные изменения в пубертатный период или в период беременности могут спровоцировать увеличение размера нейрофибром.
Нейрофиброматоз не является противопоказанием к прививкам, проведению массажа, нахождению ребенка на солнце, ЛФК. За исключением случаев, когда указанные процедуры являются противопоказанием при наличии сопутствующего заболевания.
Узнать больше о развитии детей с нейрофиброматозом 1 типа вы можете здесь.
Нф1 что это диагноз
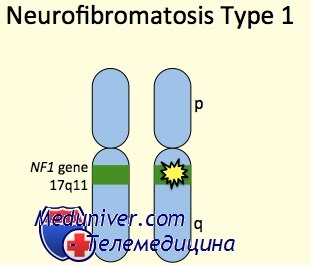
Этиология и встречаемость нейрофиброматоза I типа. Нейрофиброматоз I типа (NF1, MIM №162200) — панэтническое аутосомно-доминантное заболевание с симптомами, чаще проявляющимися в коже, глазах, скелете и нервной системе. NF1 вызван мутациями в гене нейрофибромина (NF1). Болезнь встречается с частотой 1 на 3500 человек, что делает ее одним из наиболее частых аутосомно-доминантных заболеваний.
Приблизительно половина пациентов имеет новые мутации; частота мутаций в гене NF1 — одна из самых высоких среди всех генов человека, приблизительно 1 мутация на 10 000 живых новорожденных. Около 80% новых мутаций отцовского происхождения, однако влияние отцовского возраста на частоту мутации не подтверждено.
Патогенез нейрофиброматоза I типа
NF1 — большой ген (350 килобайт, 60 экзонов), кодирующий нейрофибромин, белок, широко экспрессируемый почти во всех тканях, но наиболее обильно в головном и спинном мозге и периферической нервной системе. Считают, что нейрофибромин регулирует несколько внутриклеточных процессов, в том числе активацию ГТФазы Ras, управляя клеточным делением и функционируя как супрессор опухолевого роста.
В гене NF1 описано более 500 мутаций, большинство из которых уникальны для каждой семьи. Клинические проявления связаны с потерей функции продукта гена; 80% мутаций вызывает укорочение молекулы белка. Мутация как причина болезни может быть выявлена более чем у 95% больных нейрофиброматозом I типа.
Нейрофиброматоз I типа характеризуется крайней клинической изменчивостью, как меж-, так внутрисемейной. Эта изменчивость, вероятно, вызвана комбинацией генетических, негенетических и случайных факторов. Отчетливой корреляции генотип-фенотип не обнаружено, хотя большие делеции чаще наблюдают у больных с неврологическими проблемами.
Фенотип и развитие нейрофиброматоза I типа
Нейрофиброматоз I типа — мультисистемное заболевание с неврологическими, мышечно-скелетными, глазными и кожными аномалиями, а также предрасположенностью к новообразованиям. Диагноз нейрофиброматоз I типа может быть поставлен, если обнаружены два или более следующих критериев: шесть или более пятен цвета «кофе с молоком», имеющие в диаметре, по крайней мере, 5 мм (до пубертата) или 15 мм (после пубертата); две или больше нейрофибромы любого типа или одну плексиформную нейрофиброму; веснушки в подмышечной или паховой области; глиому зрительного нерва; два или более узелков Лиша; характерный костный фенотип (сфеноидная дисплазия и уменьшение коры длинных трубчатых костей как с, так и без псевдоартрозов); наличие родственника первой степени родства с нейрофиброматозом I типа.
Почти все больные с нейрофиброматозом I типа без семейного анамнеза не имеют клинических критериев ранее 8 лет. Детям, унаследовавшим нейрофиброматоз I типа, диагноз обычно может быть установлен клинически уже на первом году жизни, так как для них требуется наличие только одного симптома болезни.
Хотя пенетрантность по существу полная, проявления чрезвычайно вариабельны. Множество пятен цвета «кофе с молоком» присутствуют почти у всех больных, веснушки наблюдают в 90% случаев. Множество больных с нейрофиброматозом I типа имеют только кожные симптомы болезни и узелки Лиша на радужке.
Многочисленные нейрофибромы обычно присутствуют у взрослых. Плексиформные нейрофибромы встречаются реже. Глазные проявления включают глиомы зрительного нерва (могут вести к слепоте) и узелки Лиша на радужке. Наиболее тяжелые осложнения со стороны костной системы — сколиоз, дисплазия позвонков, псевдоартрозы и избыточный рост. Часто встречаются также стеноз легочных, почечных и церебральных сосудов и гипертония. Наиболее частые опухоли у детей с нейрофиброматозом I типа (кроме нейрофибром) — глиомы зрительного нерва, опухоли мозга и злокачественные миелоидные болезни.
До половины всех детей с нейрофиброматозом I типа имеют когнитивные проблемы или дефицит внимания, сохраняющиеся во взрослом возрасте.
У больных с симптомами нейрофиброматоза I типа, ограниченными одной областью тела, имеющих здоровых родителей, может диагностироваться сегментный (или локальный) нейрофиброматоз I типа. Сегментный нейрофиброматоз I типа может представлять случайное необычное распределение клинических симптомов или соматический мозаицизм по мутации в гене NF1.
Особенности фенотипических проявлений нейрофиброматоза I типа:
• Возраст начала: от пренатального до позднего детства
• Пятна «кофе с молоком»
• Веснушки в подмышечных и паховых областях
• Нейрофибромы
• Узелки Лиша (гамартомы радужки)
• Плексиформные нейрофибромы
• Глиомы зрительного нерва
• Специфические повреждения костей
Лечение нейрофиброматоза I типа
Нейрофиброматоз I типа — клинический диагноз. Идентификацию мутаций в настоящее время стандартно не проводят из-за большого размера гена и крайней аллельной гетерогенности.
Эффективного лечения нет, следовательно, помощь сфокусирована на симптоматической терапии. Постоянное наблюдение пациентов с нейрофиброматозом I типа должно включать ежегодный медицинский осмотр, проводимый знакомым с этим заболеванием врачом, ежегодный осмотр окулиста в детстве (реже у взрослых), регулярную оценку психомоторного развития в детстве и регулярный контроль артериального давления.
Деформации, вызываемые нейрофиброматозом I типа, — наиболее беспокоящее проявление болезни. Отдельные кожные и подкожные нейрофибромы можно удалить хирургическим способом, если они доставляют косметические или другие неудобства. Плексиформные нейрофибромы, вызывающие обезображивание или беспокойства, также могут быть удалены хирургически. Тем не менее хирургическое вмешательство для этих неоплазм может быть проблематичным, так как они часто интимно спаяны с нервами и имеют тенденцию рецидивировать в месте резекции.
Риски наследования нейрофиброматоза I типа
Пациенты с нейрофиброматозом I типа имеют 50% риска родить ребенка, пораженного болезнью, хотя симптоматика у ребенка может отличаться от имеющейся в семье. Пренатальная диагностика доступна в семьях с известной мутацией в гене NF1, вызывающей болезнь, или информативных по сцеплению. Хотя пренатальный диагноз точен, он не обеспечивает прогностическую информацию из-за крайней фенотипической изменчивости болезни.
Родители больного ребенка, не имеющие никаких признаков болезни, имеют небольшое повышение риска повторения при следующей беременности из-за возможности полового мозаицизма, подтвержденного при нейрофиброматозе I типа.
Пример нейрофиброматоза I типа. Л.М., 2-летний мальчик, проходит обследование в связи обнаружением пяти пятен цвета «кофе с молоком», три из них более 5 мм в диаметре. У него не найдено веснушек в подмышечных или паховых областях, пороков развития кости и нейрофибром. Клинический осмотр обоих родителей не выявил признаков нейрофиброматоза. Генетик сообщил родителям и направившему педиатру, что не обнаружил клинических критериев для нейрофиброматоза I типа.
Ребенок повторно обратился в клинику генетики в 5 лет. У него появились узелки Лиша на радужке глаз и 12 пятен цвета «кофе с молоком», 8 из которых превышали 5 мм в диаметре. Обнаружена также веснушчатость в подмышечных областях с двух сторон. Установлен диагноз нейрофиброматоз I типа; родителям сообщили, что у ребенка новая мутация, следовательно, риск повторения низкий, но не исключен гонадный мозаицизм.
Родители отказались как от молекулярных анализов, так и от пренатальной диагностики при последующей беременности.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Нф1 что это диагноз
Нейрокутанные синдромы — группа гетерогенных заболеваний с сочетанным поражением кожи и ЦНС. Большинство заболеваний имеет наследственный характер и, вероятно, развивается в результате нарушения дифференциации примитивной эктодермы. К группе нейрокутанных синдромов относятся нейрофиброматоз, туберозный склероз, болезнь Стерджа-Вебера, болезнь Клиппеля-Линдау, атаксия-телеангиэктазия, синдром линейного невуса, гипомеланоз Ито и недержание пигмента.
Нейрофиброматоз (НФ), или болезнь Реклингхаузена, — распространенное заболевание с аутосомно-доминантным типом наследования. Оно характеризуется полиморфизмом клинических проявлений, так как в процесс могут вовлекаться практически все системы и органы, и прогрессирующим течением: характерные признаки заболевания могут присутствовать уже при рождении, однако развитие осложнений может быть отсрочено на несколько десятилетий. НФ возникает в результате нарушения дифференциации клеток нервного гребня и процессов миграции на ранних стадиях эмбриогенеза.
Выделяют две различные формы нейрофиброматоза (НФ). Наиболее распространен нейрофиброматоз типа 1 (НФ-1), встречающийся с частотой 1:4000. Диагноз ставится при наличии любых двух из следующих признаков.
1. Шесть пятен цвета кофе с молоком или более размером более 5 мм до полового созревания и более 15 мм после полового созревания. Пятна цвета кофе с молоком — отличительный признак заболевания, они присутствуют практически у 100 % пациентов с НФ, зачастую уже при рождении, но затем их размер и количество увеличиваются, пигментация становится более выраженной, особенно в первые годы жизни. Пятна могут располагаться на разных участках кожи, преимущественно на туловище и конечностях, пятна на лице менее характерны.
2. Веснушчатость <точечная гиперпигментция) в подмышечной или паховой областях — множественные гиперпигментированные участки размером 2-3 мм в диаметре.
3. Два узелка Лиша на радужке или более. Узелки Лиша — гамартомы, локализованные на радужку лучше выявляются при исследовании с помощью щелевой лампы. Они определяются более чем у 74 % пациентов с НФ-1, но нехарактерны для НФ-2. Вероятность этого симптома увеличивается с возрастом; узелки Лиша определяются только у 5 % пациентов до 3 лет, у 42 % детей в возрасте 3-4 лет и у 100 % взрослых в возрасте 21 год или старше.
4. Две нейрофибромы или более либо одна плексиформная нейрофиброма. Нейрофибромы в типичных случаях возникают на коже, но могут располагаться вдоль стволов периферических нервов и кровеносных сосудов, а также во внутренних органах, включая ЖКТ. Кожные нейрофибромы выявляются, как правило, в подростковом возрасте или во время беременности, что предполагает влияние гормональных факторов. Они обычно небольшого размера, упругой, эластичной консистенции, с легким багрянистым (фиолетовым) оттенком окружающей кожи.
Плексиформные нейрофибромы обычно выявляются при рождении и возникают в результате диффузного утолщения нервных стволов, которые часто локализуются на лице в орбитальной или височной области. Кожа вокруг плексиформной нейрофибромы может быть гиперпигментирована и выглядит темнее, чем пятна цвета кофе с молоком. Плексиформная нейрофиброма может вызывать чрезмерный рост конечности и деформацию соответствующей кости.
5. Изменения костей. Сколиоз представляет собой одно из наиболее распространенных ортопедических нарушений при НФ-1, хотя это менее специфичный симптом, поэтому он не относится к диагностическим критериям. 6. Глиома зрительного нерва бывает примерно у 15 % пациентов с Нф-I. Эта относительно доброкачественная опухоль состоит из глиозных клеток и муцинозного вещества.
У большинства пациентов с глиомой зрительного нерва клинические проявления отсутствуют; зрение нормальное или почти нормальное, но примерно у 20 % из них выявляются зрительные нарушения или признаки преждевременного полового развития вследствие опухолевой инвазии гипоталамуса. Дети редко ощущают одностороннюю потерю зрения, что может служить причиной поздней диагностики заболевания. У пациентов с односторонней глиомой зрительного нерва как правило нарушена реакция зрачка на свет. Для исследования реакции зрачка на свет каждый глаз поочередно стимулируют ярким светом. На стороне поражения зрачок не сужается, но при освещении здорового глаза возникает равномерное сужение зрачков с двух сторон.
У пациентов с нейрофиброматоза (НФ)-1 и плексиформной невромой века высок риск глиомы зрительного нерва на стороне нейрофибромы. Признаками глиомы зрительного нерва на МРТ служит диффузное утолщение, локальное увеличение или ограниченный объемный процесс, локализованный в области зрительного нерва или хиазмы.
7. У пациента имеются родственники первой степени родства с НФ-1, диагноз которого базируется на приведенных выше критериях. Большинство мутаций при НФ-1 наследуется по отцовской линии. Ген НФ-1 локализован на хромосоме 17qll.2 и кодирует все мРНК размером 11-13 kb, содержащие по крайней мере 59 экзонов, которые продуцируют белок нейрофибрин.
У детей с НФ-1 имеется риск поражения нервной системы. MP-исследование у отдельных детей выявило патологические сигналы в области бледного шара, таламуса и внутренней капсулы, что может быть признаком низкодифференцированной глиомы или гамартомы, не выявляемой на КТ. Эти изменения могут быть ассоциированы с частым снижением школьной успеваемости, синдромом дефицита внимания и речевых нарушений у детей с НФ-1. Сложные парциальные и генерализованные тонико-клонические приступы — частое осложнение заболевания.
Гидроцефалия — редкий симптом, возникающий вследствие стеноза водопровода мозга, макроцефалия с нормальным размером желудочков встречается часто. Возможно поражение сосудов мозга с развитием аневризмы или стеноза вследствие болезни моя-моя. Возможны неврологические нарушения, включая транзиторные ишемические атаки, гемипарез, и когнитивные нарушения. Неудивительно, что выраженность психических нарушений зависит от тяжести заболевания и полиморфизма клинической картины. Признаки преждевременного полового созревания могут быть выражены как в ассоциации с поражением хиазмы зрительных нервов и гипоталамуса, так и при его отсутствии.
Злокачественные опухоли также относятся к тяжелым осложнениям НФ-1. Нейрофиброма в некоторых случаях может перерождаться в нейрофибросаркому или злокачественную шванному. У пациентов с НФ-1 высок риск гипертензии в результате стеноза сосудов почек или феохромоцитомы. Частота развития феохромоцитомы, рабдомиосаркомы, лейкоза, опухоли Вильмса выше у пациентов с НФ-1, чем в общей популяции. Описано необычное сочетание миелоидного лейкоза, ювенильной ксантогранулемы и НФ-1. Опухоли ЦНС, включая глиому зрительных нервов, менингиому ствола и спинного мозга, нейрофиброму, астроцитому и невриному, ответственны за значительную морбидность и летальность в связи с повышением их распространенности у пациентов с НФ-1.
Тип 2 диагностируется примерно у 10 % пациентов с нейрофиброматозом (НФ), встречается с частотой 1:50 000. Диагноз может быть установлен при наличии хотя бы одного из следующих признаков:
1) билатеральные новообразования VIII черепного нерва, представляющие собой невриномы слухового (VIII) нерва, выявляемые на КТ или МРТ;
2) наличие родственников первой степени родства (родителей, детей, сиблингов) с НФ-2 в сочетании с унилатеральным новообразованием VIII черепного нерва или любыми двумя из следующих признаков: нейрофиброма, менингиома, глиома, шваннома или ювенильное заднее подкапсульное помутнение хрусталика.
Билатеральная невринома слухового нерва — основной отличительный симптом НФ-2. Нарушение слуха, слабость мимических мышц, головная боль и нарушение координации могут появиться в детском возрасте, признаки объемного процесса в мостомозжечковом углу чаще выявляются на 2-м или 3-м десятилетии жизни. Хотя пятна цвета кофе с молоком и кожные нейрофибромы служат классическим проявлением НФ-1, они значительно реже встречаются при НФ-2. Заднее подкапсульное помутнение хрусталика выявляется примерно у 50 % пациентов с НФ-2. Как и при НФ-1, опухоли ЦНС, включая шванномы, глиальные опухоли и менингиомы, часто встречаются у пациентов с НФ-2. Ген НФ-2, по данным хромосомного анализа, локализован рядом с центром длинного плеча хромосомы 22ql.ll.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
НЕЙРОФИБРОМАТОЗ I ТИПА. Проблемы диагностики и лечения
Каковы диагностические критерии нейрофиброматоза? Что является особенностью нейрофиброматоза и затрудняет диагностику? На чем основывается терапия нейрофиброматоза? Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингха
Каковы диагностические критерии нейрофиброматоза?
Что является особенностью нейрофиброматоза и затрудняет диагностику?
На чем основывается терапия нейрофиброматоза?
 Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Заболевание характеризуется выраженным клиническим полиморфизмом, прогрессирующим течением, полиорганностью поражений и высокой частотой осложнений, в том числе приводящих к летальному исходу (развитие сердечно-легочной недостаточности вследствие выраженных скелетных аномалий, злокачественное перерождение нейрофибром и др.).
Механизм развития клинических проявлений неизвестен. Существует предположение, что ген «нф 1» входит в группу генов, подавляющих рост опухолей. Снижение или отсутствие выработки продукта гена — нейрофибромина приводит к диспластической или неопластической пролиферации клеток.
Клиническая диагностика нейрофиброматоза I типа основывается на обнаружении диагностических критериев, рекомендованных Международным комитетом экспертов по нейрофиброматозу. Диагноз может быть поставлен при наличии у больного по крайней мере двух из перечисленных ниже признаков: не менее пяти пятен цвета «кофе с молоком» диаметром более 5 мм у детей препубертатного возраста и не менее шести таких пятен диаметром более 15 мм в постпубертатном периоде; две и более нейрофибромы любого типа или одна плексиформная нейрофиброма; множественные мелкие пигментные пятна типа веснушек, локализованные в крупных кожных складках (подмышечных и/или паховых); глиома зрительного нерва; два и более узелков Лиша на радужной оболочке, обнаруживаемых при исследовании с помощью щелевой лампы; дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных трубчатых костей с наличием псевдоартроза или без него; наличие у родственников первой степени родства нейрофиброматоза I типа по тем же критериям.
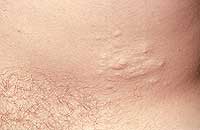
 |
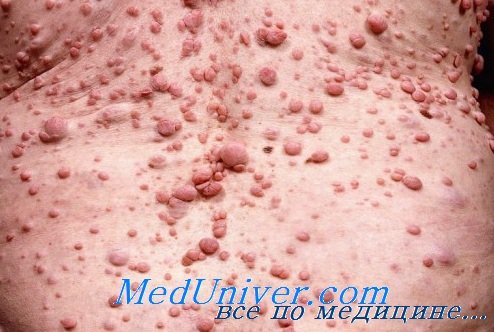
| Рисунок 1. Множественные нейрофибромы |
Особенностью заболевания является специфическая последовательность проявления симптомов в зависимости от возраста пациента, что затрудняет клиническую диагностику нейрофиброматоза I типа в раннем детском возрасте. Таким образом, с рождения или первых лет жизни могут существовать лишь некоторые признаки нейрофиброматоза I типа, такие как крупные пигментные пятна, плексиформные нейрофибромы, скелетные дисплазии. Другие симптомы могут проявиться значительно позднее (к 5–15 годам). При этом степень выраженности клинических проявлений, течение и быстрота прогрессирования нейрофиброматоза I типа у разных больных неодинаковы и колеблются в широких пределах. В настоящее время не установлено, чем обусловлены такие различия.
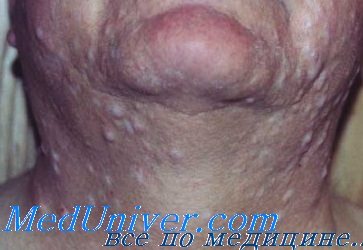
Нейрофибромы (дермальные, гиподермальные, плексиформные) представляют собой наиболее выраженное проявление болезни Реклингхаузена, их количество иногда достигает нескольких тысяч; плексиформные нейрофибромы могут быть гигантскими, массой более 10 кг. Эти косметические дефекты, как правило, больше всего беспокоят пациентов, даже имеющих системные заболевания. Кроме того, нейрофибромы, особенно плексиформные, связаны с повышенным риском озлокачествления (в 20% случаев, по нашим данным). При локализации в средостении, в брюшной полости, в глазнице они приводят к нарушению функций прилегающих органов. Например, в сентябре 2000 года в отделении наследственных заболеваний кожи ЦНИКВИ был консультирован больной мальчик 8 лет, прибывший из Брянской области в Москву для оперативного лечения по жизненным показаниям; гигантская плексиформная нейрофиброма располагалась в верхнем средостении, деформировала нижнюю 1/3 шеи и являлась причиной затрудненного дыхания и пароксизмальной тахиаритмии.
О развитии нейрофибром известно немногое. Время от времени растет их количество и размеры в ответ на различные стимулы, среди которых ведущее место занимают гормональная перестройка организма: пубертатный возраст, период беременности или после родов, а также перенесенные травмы или тяжелые соматические заболевания. С расширением спектра предлагаемых коммерческих медицинских и косметических услуг значительно увеличилось число обращений больных, указывающих на появление новых опухолей (нейрофибром, неврином, шванном) после ятрогенных вмешательств. Речь идет об удалении опухолей с диагностической или лечебной целью различными методами, в том числе с помощью хирургического иссечения. К ятрогенным осложнениям также приводит назначение физиотерапевтических процедур при лечении различных соматических заболеваний, коррекции скелетных нарушений (всевозможных видов сколиоза, переломов) и нервно-мышечных расстройств (очень часто массаж по различным поводам назначается детям грудного возраста, когда диагностика нейрофиброматоза I типа зачастую невозможна из-за недостаточности клинических проявлений). Но часто заболевание прогрессирует и на фоне кажущегося благополучия. Положение осложняется тем, что у врачей нет возможностей приостановить развитие болезни.
Основной задачей научных исследований представляется разработка методов патогенетического лечения нейрофиброматоза I типа, позволяющих сдерживать появление новых и рост уже имеющихся опухолей, а также предотвращать развитие осложнений.
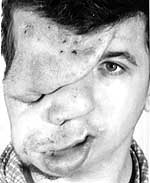 |
| Рисунок 2. Плексиформная нейрофиброма |
В настоящее время для лечения этого заболевания как за рубежом, так и в России используются методы симптоматической терапии, как, например, хирургическое удаление опухолей, коррекция кифосколиоза или лучевая терапия нейрофибром внутренних органов. Кроме того, ученые на Западе сконцентрировались на возможности этиологического лечения, то есть генной инженерии. Особенно далеко это направление продвинулось со времени открытия мутантного гена и расшифровки его первичного продукта — нейрофибромина в 1990 году; на научные изыскания, связанные с этой проблемой, ежегодно выделяются огромные средства.
Первая попытка патогенетического подхода к лечению была сделана V. Riccardi в 1987 г., когда он предложил длительное использование кетотифена (в дозе 2-4 мг в течение 1,5-3 лет) для стабилизации мембран тучных клеток, полагая, что именно дегрануляция этих клеток стимулирует рост опухолей. Однако лечение одним кетотифеном не принесло желаемых результатов: уменьшались субъективные ощущения болезненности и зуда в области нейрофибром, но какого-либо влияния на рост опухолей отмечено не было. Кроме того, при продолжительном приеме препарата наблюдалось снижение количества иммунокомпетентных клеток в периферической крови и ухудшение показателей иммунитета. Существуют различные точки зрения на роль тканевых базофилов в развитии нейрофибром. По мнению некоторых авторов, тучные клетки представляют собой эффекторные клетки противоопухолевого иммунитета. С помощью обычной и электронной микроскопии было показано, что большое количество тканевых базофилов с их активной внеклеточной дегрануляцией наблюдается только на ранней стадии развития нейрофибром. На поздней же стадии, при длительности существования нейрофибром не менее пяти лет, в клеточном матриксе опухолей тучных клеток значительно меньше, дегрануляция их преимущественно внутриклеточная и не сопровождается разрушением клеток.
На основании данных многочисленных исследований нами впервые разработана комплексная методика патогенетической терапии с использованием препаратов из разных групп лекарственных средств. Учитывая то, что клеточный состав нейрофибром в основном представлен шванновскими клетками, фибробластами, тучными клетками и лимфоцитами, а межклеточное вещество в активно растущих, особенно плексиформных, — кислыми мукополисахаридами, для лечения нейрофиброматоза I типа мы выбрали следующие препараты. Стабилизатор мембран тучных клеток, кетотифен, мы назначали по 2-4 мг короткими курсами по два месяца. Чтобы избежать осложнений, в первые две недели приема препарата использовался фенкарол по 10-25 мг три раза в день. В качестве антипролиферативного препарата применялись тигазон в дозе не менее 1 мг на килограмм массы тела или аевит до 600 000 МЕ с учетом переносимости. Также курсами применялась лидаза (мукополисахаридаза) в дозе 32-64 Ед в зависимости от возраста внутримышечно, через день, на курс 30 инъекций.
Вышеуказанные препараты использовались комплексно в различных сочетаниях или в виде монотерапии в зависимости от формы нейрофиброматоза I типа, жалоб, течения, а также возраста и пола больных. Обязательно лечение проводилось в периоды прогрессирования заболевания, то есть при появлении новых опухолей и/или росте уже имеющихся, как правило сопровождающемся зудом или ощущением болезненности в их проекции, а также с целью предотвращения активизации заболевания во время планируемых операций на опухолях. Повторные курсы лечения с интервалами в два месяца назначались при наличии у больных крупных плексиформных нейрофибром или болезненных неврином. При этом, как правило, курсовое применение тигазона (или аевита) в виде монотерапии чередовалось с сочетанным применением стабилизаторов мембран тучных клеток и инъекций лидазы. Предлагаемое лечение хорошо переносилось больными.
В единичных случаях наблюдались незначительное повышение уровней печеночных показателей в повторных биохимических анализах крови при приеме тигазона (у одной больной) и очаговая аллергическая реакция на введение лидазы (у двух пациентов из 60), которая проявлялась воспалением тканей в месте инъекций. В этих случаях препараты отменялись, назначалось симптоматическое лечение.
В результате проводимой терапии нам, как правило, удавалось приостановить прогрессирование заболевания; наблюдалось уменьшение (сморщивание) нейрофибром и неврином вплоть до полного исчезновения некоторых опухолей (особенно активно уменьшаются плексиформные нейрофибромы — на ранней стадии своего развития — и невриномы).
Полученные результаты удовлетворяют исследователей и позволяют рекомендовать вышеуказанную методику патогенетического лечения нейрофиброматоза I типа для повсеместного применения. Разработанная нами комплексная методика патогенетического лечения впервые дает возможность оказать больным медикаментозную помощь.


