Синдром Жильбера
| молекулярно-генетический анализ промоторной области гена UGT1A1 , срок 8 р. дней UAH | Записаться |
 Недостаточность фермента при синдроме Жильбера приводит к незначительному повышению билирубина в крови, особенно после потребления алкоголя, скудном питании или обезвоживании. Периодически это приводит к возникновению умеренной желтухи – кожный покров, белки глаз, слизистые оболочки окрашиваются в желтый цвет.
Недостаточность фермента при синдроме Жильбера приводит к незначительному повышению билирубина в крови, особенно после потребления алкоголя, скудном питании или обезвоживании. Периодически это приводит к возникновению умеренной желтухи – кожный покров, белки глаз, слизистые оболочки окрашиваются в желтый цвет.
Генетическая диагностика синдрома Жильбера
Синдром Жильбера обусловлен мутацией гена UGT1A1, кодирующего фермент глюкуронилтрансферазу. Этот ген находится на хромосоме 2 (2q37). ДНК-диагностика синдрома Жильбера проводится путем анализа числа ТА-повторов в промоторной области гена UGT1A1. В норме повторов 6. При синдроме Жильбера их больше 7 в обеих хромосомах: A(TA)7TAA / A(TA)7TAA.
Данный тест — самый быстрый и эффективный способ дифференциальной диагностики болезни с печеночными поражениями, сопровождающимися гипербилирубинемией, а также профилактики печеночных кризов, изменения образа жизни больного. Генетические анализы на синдром Жильбера рекомендуют сдавать пациентам, которым предстоит прием препаратов с гепатотоксическим эффектом.
Другие мутации этого же гена приводят к синдрому Криглера-Найара, который является более опасной формой гипербилирубинемии (высокого уровня билирубина в крови).
Ген UGT1A1 входит в Панель «Наследственные заболевания желудочно-кишечного тракта»
Как выглядит результат генетического анализа на синдром Жильбера
Высокий риск развития Синдрома Жильбера. 
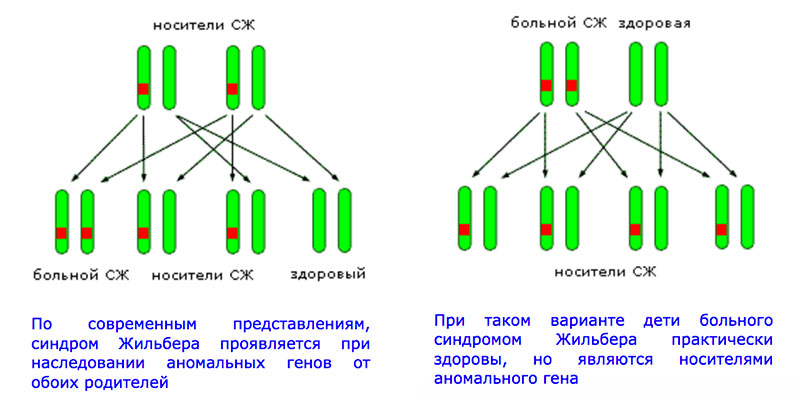
Как наследуется анормальный ген
Многие люди являются носителями анормального гена UGT1A1. Для возникновения синдрома Жильбера у ребенка, у него должны быть 2 копии гена с поломкой в промоторной области, то есть носителями аномального гена должны быть оба родителя. Синдром Жильбера наследуется по аутосомно-рецессивному типу.
Факторы риска синдрома Жильбера
Хотя синдром Жильбера присутствует с детства, он остается обычно незамеченным до подросткового возраста, поскольку выработка билирубина увеличивается во время полового созревания. Болезнь обнаруживается обычно случайно во время анализа крови. Болезнь имеет распространение 3%-10% в зависимости от популяции.
Симптомы синдрома Жильбера
 Наиболее характерными симптомами синдрома Жильбера считаются:
Наиболее характерными симптомами синдрома Жильбера считаются:
— дискомфортные ощущения в правом подреберье,
— достаточно быстрая утомляемость,
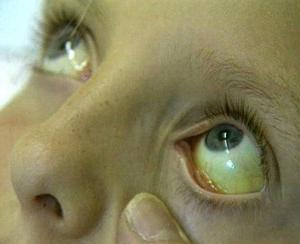
— субиктеричность и иктеричность склер (пожелтение белков глаз) и кожи,
— ксантелазмы (плоские жировые бляшки) век
— в некоторых случаях наблюдается увеличение размеров печени.
Как организм в норме обрабатывает билирубин
Билирубин проходит через печень в кишечник с желчью, а затем выделяется с калом. Небольшое количество билирубина остается в крови.
Осложнения
Низкий уровень фермента, перерабатывающего билирубин, может также увеличивать побочные эффекты некоторых медикаментов, поскольку этот фермент играет роль в выведении этих медикаментов из организма.
При синдроме Жильбера, перед приемом медикаментов следует посоветоваться с врачом. Также, некоторые виды синдрома Жильбера могут увеличивать риск развития желчно-каменной болезни.
Диагностика
При подозрении синдрома Жильбера из-за необъяснимой желтушности или повышенного уровня билирубина, доктор осматривает пациента и опрашивает про симптомы, такие как боль в брюшной полости или темная моча. Врач может рекомендовать биохимический анализ крови и анализ функции печени для исключения других проблем с печенью, которые могут быть причиной повышения билирубина.
Комбинация нормальных анализов крови, нормальных функциональных анализов печени и повышенного уровня билирубина является индикатором синдрома Жильбера. Генетическое тестирование может подтвердить диагноз.
Лечение
Синдром Жильбера обычно не требует лечения. Уровень билирубина в крови может колебаться время от времени, и иногда может возникать желтушность, которая безболезненно проходит сама по себе.
В период обострения синдрома пациенту необходимо строго соблюдать стол №5. Его суть заключается в отказе от жирных, жареных, высококалорийных и других вредных блюд. Также назначается ряд медикаментозных препаратов в виде микросомальных ферментов – Зиксорина и Фенобарбитала. Они назначаются двумя курсами по 1-2 недели с перерывом в месяц. Благодаря этим препаратам удается быстро снизить билирубин.
Образ жизни и профилактика
Синдром Жильбера при беременности
Синдром Жильбера не запрещает женщине выносить и родить ребенка. Если такое генетическое отклонение есть только у одного партнера, то у детей такое заболевание будет отсутствовать. Но такой ребенок становится носителем этого гена: он никогда не столкнется с синдромом, однако его потомство может столкнуться с таким отклонением.
Как пройти исследование
Материал для исследования: Венозная кровь с ЭДТА 4 мл.
Подготовка к анализу: нет
Дни забора (приема) материала: в часы работы медицинского центра.
Генетическая диагностика синдрома Жильбера, UGT 1A1
Генетическая диагностика синдрома Жильбера заключается в анализе количества повторов динуклеотидной последовательности тимидин-аденин (ТА)- гена, кодирующего фермент УДФГТ или уридиндифосфатглюкуронидазу, который отвечает за превращение молекулы билирубина в водорастворимую форму. Место расположения гена – хромосома 2. Мутировавший участок гена приводит не только к формированию синдрома Жильбера, но и провоцирует синдром Криглера-Найяра (описано ниже), преходящую желтуху новорожденных, повышенный сывороточный билирубин.
Описание
Характерен астенический синдром с вегетативными расстройствами – утомляемость, слабость, нарушения засыпания, потливость, дискомфорт в сердце. Если повышенный уровень билирубина держится долго, развивается депрессия.
Уровень билирубина повышается изолированно, чаще до 100 ммоль/л, преобладает прямая (несвязанная) фракция, остальные печеночные пробы не изменяются.?
Механизм развития желтухи следующий. В результате распада эритроцитов образуется прямой билирубин, он циркулирует в крови. В норме в клетках печени прямой билирубин захватывается глюкуроновой кислотой и становится непрямым. «Командует» связыванием прямого билирубина фермент УДФГТ, активность которого при синдроме Жильбера снижена на 25-30%. Билирубин, оставшийся несвязанным, накапливается в тканях, вызывая их повреждение. Лечение симптоматическое – диета №5 (ограничение жирного, жареного, содержащего холестерин, много овощей и фруктов), покой, по назначению врача гепатопротекторы.
Фермент УДФГТ синтезируется по коду, который задает ген UGT 1A1. Синдром Жильбера – это мутация промоторной области (участок нуклеотидов, с которого начинается кодирующая часть), вместо 6 ТА-повторов обнаруживается 7 или реже 8.
Мутация – это изменение генотипа, которое передается потомкам. Генотип – это набор генов, который уникален и неповторим у каждого человека. Мутация гена UGT 1A1 передается по аутосомно-доминантному типу. Это значит, что дефектный ген не содержится в половых хромосомах, мог быть передан как от отца, так и от матери. Дети обоих полов могут болеть с одинаковой частотой.
Имеет значение тип мутации – гомо- или гетерозиготная. Нужно вспомнить, что каждый организм имеет два набора хромосом – от отца и от матери. Гомозиготная мутация (в переводе с греческого «равная, одинаковая яйцеклетка») – такая, при которой пара хромосом имеет одинаковую форму гена. Гетерозиготная мутация отличается тем, что форма генов в паре разная.
При гомозиготной мутации или одинаковой паре генов клиническая картина выражена тяжелее, уровень билирубина максимально высокий. При гетерозиготной форме повышение билирубина обнаруживается случайно, клинических проявлений нет. Бессимптомное течение отмечается в более чем половине случаев синдрома Жильбера.
В группе риска находятся все люди, у которых есть родственники с синдромом Жильбера. Вылечить его невозможно, нужно ограничивать нагрузки на печень, избегать провоцирующих факторов. Продолжительности жизни синдром Жильбера не уменьшает, влияние на качество жизни минимальное. Это особенность организма, с которой нужно считаться, чтобы не навредить себе.
Определять наличие мутантного гена нужно перед началом лечения препаратами, оказывающими токсическое действие. У людей с синдромом Жильбера могут быть тяжелые интоксикационные реакции при приеме многих групп лекарств: глюкокортикоидных и половых гормонов, кофеина, некоторых антибиотиков, аспирина, парацетамола. Особенно это касается приема нового цитостатика Иринотекана, чрезвычайно эффективного при раке толстой кишки.
Специалисты ЦЭЛТ предпочитают полностью обследовать пациента перед сложным лечением во избежание неприятных неожиданностей.
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, врача общей практики со стажем в 14 лет.

Определение болезни. Причины заболевания
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
К провоцирующим факторам проявления синдрома можно отнести:
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
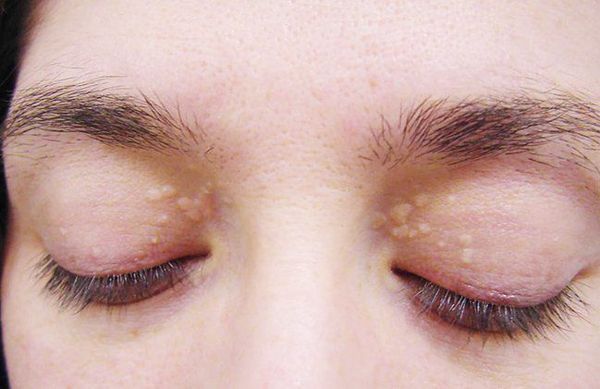
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
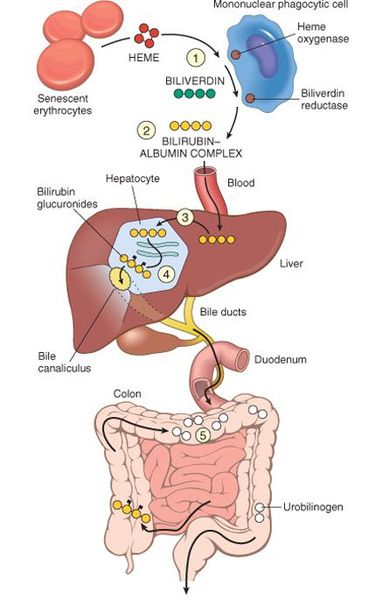
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
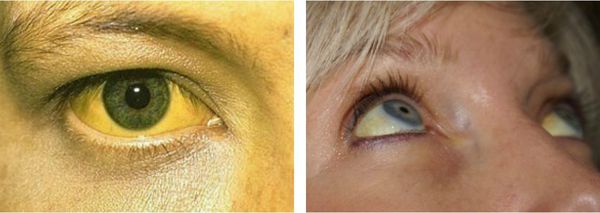
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами («жирами») клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
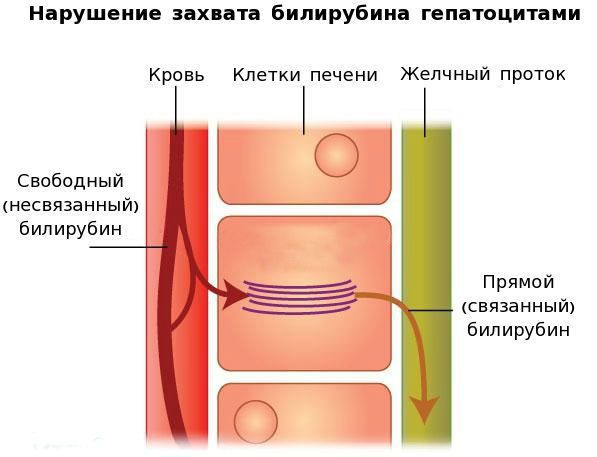
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Обнаружена гомозиготная мутация что это значит у женщин
Молекулярно-генетическая диагностика синдрома Жильбера, основанная на исследовании промоторной области гена UGT1A1. Синдром Жильбера – доброкачественная неконъюгированная гипербилирубинемия умеренной выраженности.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Подробнее об исследовании
Синдром Жильбера – это наследственное заболевание, проявляющееся эпизодами желтухи и повышением уровня неконъюгированного (свободного, непрямого) билирубина в сыворотке крови. Его распространенность составляет около 5 %.
Причина развития синдрома – снижение активности фермента печени уридиндифосфат-глюкуронилтрансферазы (УДФГТ), который кодируется геном UGT 1A1. Мутация в промоторной области гена UGT 1A1 характеризуется увеличением количества ТА-повторов (в норме их число не превышает 6). Если их становится 7 (или реже 8) в гомозиготном или гетерозиготном состоянии, функциональная активность фермента УДФГТ снижается – это обязательное условие для возникновения синдрома Жильбера. У гомозиготных носителей мутации заболевание характеризуется более высоким исходным уровнем билирубина и более тяжелыми клиническими проявлениями. У гетерозиготных носителей преобладает латентная форма заболевания.
В норме при распаде эритроцитов выделяется непрямой билирубин, который необходимо вывести из организма. Поступив в клетки печени, он связывается с глюкуроновой кислотой под влиянием фермента уридиндифосфат-глюкуронилтрансферазы (УДФГТ). Соединение билирубина с глюкуроновой кислотой делает его растворимым в воде, что обеспечивает возможность его перехода в желчь и выделения с мочой. Из-за мутации в гене UGT1A1 и, как следствие, недостаточной активности УДФГТ конъюгация непрямого билирубина нарушается, что приводит к повышению его концентрации в крови. Увеличение содержания билирубина в крови, в свою очередь, способствует накоплению его в тканях, особенно в эластической ткани (содержится в стенке кровеносных сосудов, коже, склерах) – этим объясняется желтушность.
Проявления синдрома Жильбера могут возникать в любом возрасте и провоцируются физическими нагрузками, стрессовыми ситуациями, голоданием, вирусными инфекциями, приемом алкоголя, ряда лекарственных препаратов, обладающих гепатотоксическим эффектом. У заболевания неспецифические симптомы: боли в животе, тяжесть в правом подреберье, расстройства пищеварения (тошнота, отрыжка, запоры, диарея), усталость, общее недомогание, тревожность. Основной симптом – желтушное окрашивание кожи и слизистых оболочек и повышение уровня непрямого билирубина в крови. Гипербилирубинемия (повышение уровня билирубина) чаще всего может составлять не более 100 ммоль/л с преобладанием непрямой фракции. Остальные печеночные пробы, как правило, не изменены.
Под влиянием солнечного света у больных с синдромом Жильбера может отмечаться повышенная пигментация кожи.
Иногда заболевание проявляется в период новорождённости и расценивается как физиологическая желтуха новорождённых.
Возможно и постоянное бессимптомное течение, тогда синдром Жильбера может обнаруживаться при случайно выявленных отклонениях в биохимическом анализе крови (показатель билирубина).
Своевременная диагностика синдрома Жильбера позволяет отличить его от других заболеваний печени и крови, вовремя ограничить прием препаратов, обладающих гепатотоксическим действием, осуществить профилактику печеночных кризов, скорректировать образ жизни пациента до полного исчезновения дискомфорта, вызываемого гипербилирубинемией.
Самый быстрый способ выявить синдром Жильбера – прямая ДНК-диагностика, заключающаяся в определении числа TA-повторов в гене UGT1A1.
Факторы, провоцирующие обострение синдрома Жильбера:
Когда назначается исследование?
Что означают результаты?
Генетические маркеры
Анализ гена UGTA1 также используется для прогноза побочных эффектов при терапии препаратом «Иринотекан» у пациентов с онкологическими заболеваниями (колоректальным раком). Заключение выдается в соответствии с профилем исследования.
Бомба замедленного действия: чем опасно носительство мутаций у будущих родителей
Когда тест на беременность показывает заветные две полоски – это очень радостное и волнительное событие. Будущие родители готовятся к тому, что с появлением малыша их жизнь заиграет новыми красками. К сожалению, не для всех пар эти мечты воплощаются в реальность. Иногда счастье сменяется болью утраты из-за того, что беременность прерывается по непонятным причинам.
Пары, пережившие такие события, часто начинают винить себя. Женщина перебирает в памяти события из своей жизни, пытается понять, что она делала неправильно, где совершила роковую ошибку, из-за которой всё это произошло. На самом деле чаще всего никто не виноват. Одна из возможных причин прерываний беременности и рождения детей с тяжелыми заболеваниями – наследственность. Генетические мутации коварны. Некоторые из них передаются по аутосомно-рецессивному типу. Катастрофа происходит, когда в клетках организма встречаются две «неправильные» копии гена. Оба родителя могут оказаться носителями. Каждый из них здоров, потому что один ген функционирует нормально. Но есть 25% вероятность, что ребенок получит оба дефектных гена. Это высокий риск.

Еще существуют наследственные заболевания, сцепленные с полом. Тут тоже довольно сложный механизм наследования. Например, если дефектный ген находится в «женской» X-хромосоме, то носительницами могут быть только женщины, а их сыновья в 50% случаев рождаются больными, в остальных 50% случаев они здоровы и не являются носителями.
Проблема в том, что такие «неправильные» гены обычно сложно выявить. У носителей нет симптомов, и они не догадываются о рисках для потомства. Зачастую это вскрывается только после нескольких прервавшихся беременностей или рождения ребенка с тяжелым заболеванием.
Случаи из нашей практики
В 2019 году в Репробанк обратилась пара, у которой было несколько потерь беременности и безуспешных попыток ЭКО. Эти люди очень хотели завести ребенка, они прошли обследование, и после тщательного обследования врач-генетик заподозрил в паре носительство одного из генетических заболеваний, относящихся к группе митохондриальной патологии.

Генетический анализ показал, что оба партнера являются носителями мутации в гене SCO2. Эта мутация связана с тяжелым заболеванием – фатальной инфантильной митохондриальной миопатией. Эта патология вызвана нарушением функции митохондрий – «клеточных электростанций», она проявляется в виде почечной недостаточности, поражения сердечной мышцы (кардиомиопатии), выраженных дыхательных нарушений, снижения мышечного тонуса, слабости, повышенного уровня молочной кислоты в крови (лактат-ацидоз).
Другая пара обратилась к нам по поводу замершей беременности на девятом месяце. Это одно из самых страшных осложнений беременности, которого сильнее всего боятся будущие мамы. Развитие плода останавливается, и он погибает. В случае с данной парой генетический анализ показал, что оба родителя являются носителями мутации, связанной со спинальной мышечной атрофией (СМА). Это наследственное заболевание может передаваться в том числе по аутосомно-рецессивному типу и характеризуется гибелью нервных клеток, ответственных за движения. Тяжелее всего протекает младенческий тип СМА: у таких детей с рождения нарушено дыхание, они не могут нормально сосать грудь, глотать, держать головку, сидеть.
Еще одна мутация, носители которой обращались в Репробанк, была связана с наследственным поликистозом почек. При этой патологии у детей примерно 90% ткани почек замещается кистами, развивается прогрессирующая почечная недостаточность. У этой пары в анамнезе было 4 потери ребенка.
Донорские половые клетки – одно из возможных решений
Для родителей, которые являются носителями одинаковых мутаций, связанных с тяжелыми наследственными заболеваниями, есть несколько решений. Вот что об этом говорит врач-генетик Александра Борисовна Тюрина:
«У партнеров, которые являются носителями мутаций в одном и том же гене, ответственном за редкую наследственную патологию, очень высок риск родить больного ребенка. Вариантов у таких семей несколько: сделать ЭКО с поиском семейной мутации у эмбриона, обследовать плод во время беременности, сделать выбор в пользу донорского материала, выбрать усыновление или отказ от деторождения вовсе. Это так называемые «reproductive options». Этот термин можно перевести как репродуктивный выбор или репродуктивные варианты. У каждого из этих вариантов есть как преимущества, так и недостатки, но нет плохого или хорошего решения. Каждая семья, столкнувшаяся с редким наследственным заболеванием, делает приемлемый для себя выбор. Репробанк помогает подобрать подходящего донора для каждой конкретной семьи и минимизировать риск рождения ребенка с наследственным заболеванием, если пара выберет этот путь».
Паре из нашего первого примера – носителям мутации SCO2 – было предложено воспользоваться донорскими половыми клетками. Для первой беременности наши специалисты оплодотворили яйцеклетку женщины донорской спермой, а для второй беременности сперматозоиды ее партнера использовали для оплодотворения донорской яйцеклетки. Оба донора были дополнительно проверены на носительство данной мутации. Теперь эта пара растит двух здоровых малышей.
«Наши стандарты отбора доноров – одни из самых строгих в мире. Донором Репробанка становится только 1 из 300 кандидатов. Мы стремимся снизить до минимума риски возникновения генетических заболеваний. В дополнение к необходимым, согласно №107н (803н) приказу, обследованиям все наши доноры обязательно проходят генетический скрининг. Многим из них проведено полноэкзомное секвенироване экзома — исследование (прочтение) всей кодирующей белок части генома.
Как итог, около 40% доноров мы отсеиваем по причине носительства того или иного частого или очень серьезного наследственного заболевания (а в некоторых случаях и нескольких одновременно). Для данной пары мы подобрали доноров спермы и яйцеклеток, не имеющих мутации в гене SCO2».
Автандил Чоговадзе, руководитель Репробанка.
Также мы могли бы предложить этой паре воспользоваться технологией предимплантационного генетического тестирования на моногенную патологию (ПГТ-М). Этот метод помог бы получить совместные эмбрионы у супругов, протестировать их и отобрать на перенос только те, что здоровы или являются здоровыми носителями. К сожалению, у этой технологии есть ряд ограничений: высокая себестоимость, значительное время для реализации. Поэтому наши пациенты отказались от этого варианта.
Все три наших примера иллюстрируют огромную роль генетических исследований при подготовке к беременности. Всем парам, планирующим завести ребенка, стоит проконсультироваться с клиническим генетиком вне зависимости от возраста, состояния здоровья и семейной истории. Это может помочь избежать трагедии в будущем.
Все доноры половых клеток в Репробанке проходят тщательное обследование, в том числе генетический скрининг. В нашем каталоге нет носителей опасных мутаций. Тем не менее, если вы решили использовать донорский материал, вам стоит пройти генетическое обследование.


