Нуклеотидная замена в гетерозиготном состоянии что это
Замена единичного нуклеотида (или точковая мутация) в последовательности ДНК может изменять код в триплете и вызывать замену одной аминокислоты другой в продукте гена. Такие мутации называются миссенс-мутациями, поскольку они изменяют значение кодирующей последовательности гена, определяя другую аминокислоту.
При многих заболеваниях, например гемоглобинопатиях, наиболее часто обнаруживаемые мутации — миссенс-мутации.
Другие замены нуклеотидов, происходящие как в пределах, так и за пределами кодирующей последовательности гена, также могут влиять на продукт гена или создавать помехи непосредственно самому процессу транскрипции.
Множество мутаций в 5′-области промотора или 3′-нетранслируемой области гена b-глобина ведет к выраженному уменьшению количества готовой, зрелой мРНК b-глобина. На самом деле такие мутации позволили объяснить значение для экспрессии генов конкретных нуклеотидов в этих областях.
Мутации прекращающие синтез цепи
Точковые мутации последовательности ДНК, вызывающие замены нормального кодона аминокислоты на один из трех стоп-кодонов, называются нонсенс-мутациями. Поскольку при достижении стоп-кодона трансляция мРНК прекращается, мутации, преобразующие один из кодирующих кодонов экзона в стоп-кодон, прекращают на полпути трансляцию кодирующей последовательности мРНК.
Последствия мутаций преждевременного завершения трансляции двойственны. Во-первых, мРНК, несущая такую мутацию, часто неустойчива (нонсенс-распад мРНК), и трансляция оказывается невозможной. Даже если полученная мРНК достаточно стабильна для трансляции, усеченный белок обычно также неустойчив и быстро деградирует в пределах клетки.
Точковая мутация может не только создать кодон преждевременного завершения трансляции, но и уничтожить стоп-кодон, позволив продолжение трансляции до следующего стоп-кодона. Такая мутация создает белок с дополнительными аминокислотами на карбоксильном конце и может нарушать любые управляющие функции, предусматриваемые 3′-нетранслируемым участком, расположенным ниже нормального стоп-кодона.
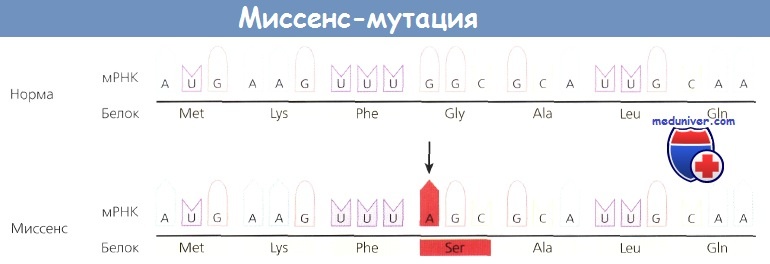 Миссенс-мутация — замена на кодон, кодирующий другую аминокислоту (может быть опасной мутацией или нейтральным полиморфизмом)
Миссенс-мутация — замена на кодон, кодирующий другую аминокислоту (может быть опасной мутацией или нейтральным полиморфизмом)
Мутации процессинга РНК
Нормальный механизм преобразования первичной РНК в зрелую мРНК требует серии модификаций, включая полиаде-нилирование, закрытие 5′-конца и сплайсинг. Созревание РНК зависит от специфических последовательностей в пределах мРНК. При сплайсинге описаны два основных класса мутаций. Чтобы получить зрелую мРНК удалением интронов и сращиванием экзонов, необходимы конкретные нуклеотидные последовательности, располагающиеся непосредственно в или около соединений экзона и интрона (5′-донорский сайт) или интрона и экзона (3′-акцепторный сайт).
Мутации, повреждающие необходимые нуклеотиды в донорском и акцепторном сайте, нарушают (и в некоторых случаях прекращают) нормальный сплайсинг РНК в этом месте.
Второй класс мутаций сплайсинга включает замены оснований в интроне, не влияющие на донорский или акцепторный сайты. Данный класс мутаций создает альтернативный донорский или акцепторный сайт, конкурирующий с нормальными сайтами в ходе сплайсинга РНК. Таким образом, в этих случаях по крайней мере часть зрелой мРНК может иметь неправильно сращенные последовательности интрона.
Горячие точки мутаций
Нуклеотидные замены, включающие замену одного пуринового основания другим (А на G или наоборот) или одного пиримидинового на другое пиримидиновое (С на Т или Т на С), называются транзициями. Замена же пуринового основания на пиримидиновое (или наоборот) называется трансверсией. Если бы нуклеотидные замены были случайными, должно было быть в два раза больше трансверсий, чем транзиций, поскольку каждое основание может подвергаться двум трансверсиям и только одной транзиций. Разные мутагенные процессы преимущественно вызывают тот или другой тип замены.
Например, среди однонуклеотидных замен, вызывающих генетические болезни, преимущественно бывают транзиций. Это наблюдение, вероятно, может объясняться тем, что основная форма модификации ДНК в геноме человека представлена метилированием остатка цитозина (с формированием 5-метилцитозина), особенно когда он располагается рядом с гуанином (т.е. как динуклеотид 5′-CG-3′).
Спонтанное деаминирование 5-метилцитозина в тимидин в CG-динуклеотиде вызывает транзицию С > Т или G > А (в зависимости от нити ДНК, в которой видоизменяется 5-метилцитозин). Более 30% всех однонуклеотидных замен относятся к этому типу, они происходят в 25 раз чаще любой другой однонуклеотидной мутации. Таким образом, CG-динуклеотид представляет настоящую «горячую точку» для мутаций в геноме человека.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Бомба замедленного действия: чем опасно носительство мутаций у будущих родителей
Когда тест на беременность показывает заветные две полоски – это очень радостное и волнительное событие. Будущие родители готовятся к тому, что с появлением малыша их жизнь заиграет новыми красками. К сожалению, не для всех пар эти мечты воплощаются в реальность. Иногда счастье сменяется болью утраты из-за того, что беременность прерывается по непонятным причинам.
Пары, пережившие такие события, часто начинают винить себя. Женщина перебирает в памяти события из своей жизни, пытается понять, что она делала неправильно, где совершила роковую ошибку, из-за которой всё это произошло. На самом деле чаще всего никто не виноват. Одна из возможных причин прерываний беременности и рождения детей с тяжелыми заболеваниями – наследственность. Генетические мутации коварны. Некоторые из них передаются по аутосомно-рецессивному типу. Катастрофа происходит, когда в клетках организма встречаются две «неправильные» копии гена. Оба родителя могут оказаться носителями. Каждый из них здоров, потому что один ген функционирует нормально. Но есть 25% вероятность, что ребенок получит оба дефектных гена. Это высокий риск.

Еще существуют наследственные заболевания, сцепленные с полом. Тут тоже довольно сложный механизм наследования. Например, если дефектный ген находится в «женской» X-хромосоме, то носительницами могут быть только женщины, а их сыновья в 50% случаев рождаются больными, в остальных 50% случаев они здоровы и не являются носителями.
Проблема в том, что такие «неправильные» гены обычно сложно выявить. У носителей нет симптомов, и они не догадываются о рисках для потомства. Зачастую это вскрывается только после нескольких прервавшихся беременностей или рождения ребенка с тяжелым заболеванием.
Случаи из нашей практики
В 2019 году в Репробанк обратилась пара, у которой было несколько потерь беременности и безуспешных попыток ЭКО. Эти люди очень хотели завести ребенка, они прошли обследование, и после тщательного обследования врач-генетик заподозрил в паре носительство одного из генетических заболеваний, относящихся к группе митохондриальной патологии.

Генетический анализ показал, что оба партнера являются носителями мутации в гене SCO2. Эта мутация связана с тяжелым заболеванием – фатальной инфантильной митохондриальной миопатией. Эта патология вызвана нарушением функции митохондрий – «клеточных электростанций», она проявляется в виде почечной недостаточности, поражения сердечной мышцы (кардиомиопатии), выраженных дыхательных нарушений, снижения мышечного тонуса, слабости, повышенного уровня молочной кислоты в крови (лактат-ацидоз).
Другая пара обратилась к нам по поводу замершей беременности на девятом месяце. Это одно из самых страшных осложнений беременности, которого сильнее всего боятся будущие мамы. Развитие плода останавливается, и он погибает. В случае с данной парой генетический анализ показал, что оба родителя являются носителями мутации, связанной со спинальной мышечной атрофией (СМА). Это наследственное заболевание может передаваться в том числе по аутосомно-рецессивному типу и характеризуется гибелью нервных клеток, ответственных за движения. Тяжелее всего протекает младенческий тип СМА: у таких детей с рождения нарушено дыхание, они не могут нормально сосать грудь, глотать, держать головку, сидеть.
Еще одна мутация, носители которой обращались в Репробанк, была связана с наследственным поликистозом почек. При этой патологии у детей примерно 90% ткани почек замещается кистами, развивается прогрессирующая почечная недостаточность. У этой пары в анамнезе было 4 потери ребенка.
Донорские половые клетки – одно из возможных решений
Для родителей, которые являются носителями одинаковых мутаций, связанных с тяжелыми наследственными заболеваниями, есть несколько решений. Вот что об этом говорит врач-генетик Александра Борисовна Тюрина:
«У партнеров, которые являются носителями мутаций в одном и том же гене, ответственном за редкую наследственную патологию, очень высок риск родить больного ребенка. Вариантов у таких семей несколько: сделать ЭКО с поиском семейной мутации у эмбриона, обследовать плод во время беременности, сделать выбор в пользу донорского материала, выбрать усыновление или отказ от деторождения вовсе. Это так называемые «reproductive options». Этот термин можно перевести как репродуктивный выбор или репродуктивные варианты. У каждого из этих вариантов есть как преимущества, так и недостатки, но нет плохого или хорошего решения. Каждая семья, столкнувшаяся с редким наследственным заболеванием, делает приемлемый для себя выбор. Репробанк помогает подобрать подходящего донора для каждой конкретной семьи и минимизировать риск рождения ребенка с наследственным заболеванием, если пара выберет этот путь».
Паре из нашего первого примера – носителям мутации SCO2 – было предложено воспользоваться донорскими половыми клетками. Для первой беременности наши специалисты оплодотворили яйцеклетку женщины донорской спермой, а для второй беременности сперматозоиды ее партнера использовали для оплодотворения донорской яйцеклетки. Оба донора были дополнительно проверены на носительство данной мутации. Теперь эта пара растит двух здоровых малышей.
«Наши стандарты отбора доноров – одни из самых строгих в мире. Донором Репробанка становится только 1 из 300 кандидатов. Мы стремимся снизить до минимума риски возникновения генетических заболеваний. В дополнение к необходимым, согласно №107н (803н) приказу, обследованиям все наши доноры обязательно проходят генетический скрининг. Многим из них проведено полноэкзомное секвенироване экзома — исследование (прочтение) всей кодирующей белок части генома.
Как итог, около 40% доноров мы отсеиваем по причине носительства того или иного частого или очень серьезного наследственного заболевания (а в некоторых случаях и нескольких одновременно). Для данной пары мы подобрали доноров спермы и яйцеклеток, не имеющих мутации в гене SCO2».
Автандил Чоговадзе, руководитель Репробанка.
Также мы могли бы предложить этой паре воспользоваться технологией предимплантационного генетического тестирования на моногенную патологию (ПГТ-М). Этот метод помог бы получить совместные эмбрионы у супругов, протестировать их и отобрать на перенос только те, что здоровы или являются здоровыми носителями. К сожалению, у этой технологии есть ряд ограничений: высокая себестоимость, значительное время для реализации. Поэтому наши пациенты отказались от этого варианта.
Все три наших примера иллюстрируют огромную роль генетических исследований при подготовке к беременности. Всем парам, планирующим завести ребенка, стоит проконсультироваться с клиническим генетиком вне зависимости от возраста, состояния здоровья и семейной истории. Это может помочь избежать трагедии в будущем.
Все доноры половых клеток в Репробанке проходят тщательное обследование, в том числе генетический скрининг. В нашем каталоге нет носителей опасных мутаций. Тем не менее, если вы решили использовать донорский материал, вам стоит пройти генетическое обследование.
Генетическое обследование

Сегодня генетическое обследование применяют в случае наличия вероятности появления какого-то генетического нарушения в семье. Данное тестирование приемлемо лишь в том случае, если структура генетического наследования нарушения достаточно изучена, возможно эффективное лечение и использованы достоверные, надежные, высокочувствительные, безвредные и специфические методики исследования. В определенном поколении преобладание должно быть весьма высоким для оправдания тех усилий, которые будут затрачены на проведение теста. Целью генетического тестирования может быть идентификация гетерозиготного носителя гена рецессивного нарушения, однако при этом не выражающего его (к примеру, у евреев ашкенази болезнь Тея-Сакса, у негров серповидно-клеточная анемия, талассемия у определенных этнических групп). Когда гетерозиготной парой выступает также гетерозигота, семья находится в зоне риска рождения нездорового ребенка.
Когда нужен тест?
Исследование может быть необходимо до того, как проявится симптоматика в том случае, когда в истории семьи была мажорирована наследовавшаяся патология, которая проявляется в более позднем возрасте (к примеру, рак молочной железы, болезнь Хантингтона). Тест определяет уровень риска развития нарушения, следовательно, человек в будущем сможет принять превентивные меры. Когда тест продемонстрировал, что человек выступает носителем нарушения, тогда он тоже может принимать решения, которые касаются рождения потомства.
Предродовой тест также может включать амниоцентез, исследование крови пуповины, взятие пробы ворсин хориона, обследование материнской крови, тест эмбрионального воплощения или материнской сыворотки. Распространенные причины для предродового обследования это:
Обследование новорожденного дает возможность осуществить профилактику (специальную диету или терапию замены) галактозного диабета, фенилпировиноградной олигофрении, а также гипотиреоза.
Также текст сегодня используют для создания семейной генеалогии. В современной генетической консультации широко используется создание семейной генеалогии (генеалогическое древо). При этом применяются условные символы, которые обозначают членов семьи и дают необходимые данные о состоянии их здоровья. Определенные семейные нарушения с похожими фенотипами обладают несколькими моделями наследования.
Митохондриальные нарушения ДНК
В митохондрии содержится уникальная округлая хромосома, которая несет информацию о тринадцати протеинах, разных РНК, а также нескольких регулятивных ферментах. Но данные о более чем 90 процентов митохондриальных протеинов есть в ядерных генах. У каждой клетки в составе есть несколько сотен митохондрий в собственной цитоплазме.
Митохондриальные нарушения часто проистекают от митохондриальных патологий или патологий ядерных ДНК (к примеру, разрушений, мутаций, дупликаций). Ткани высокой энергии (к примеру, мускулы, мозг, сердце) располагаются в зоне особенного риска нарушения функций из-за митохондриальных аномалий.
Митохондриальные патологии проявляются при множестве распространенных нарушений, к примеру, при определенных видах болезни Паркинсона (которые способны спровоцировать сильные митохондриальные делеционные мутации в тканях подкорковых узлов) и множестве других видов нарушений функционирования мышц.
Патологии митохондрии ДНК определяют наследованием со стороны матери. Митохондрии все наследуются от цитоплазмы яйцеклетки, по этой причине все потомство нездоровой матери пребывает в зоне риска наследования нарушений, однако при этом какой-либо риск наследования нарушения от больного отца отсутствует. Разнообразие клинических проявлений выступает правилом, которое способно объясняться частично вариативностью сочетаний наследованных мутаций и нормальных клеток и тканей.
Дефект одного гена
Генетические расстройства, вызванные нарушением лишь в одном гене (так называемые «менделевские нарушения»), наиболее простые для анализа и самые полно изученные на сегодняшний день. Наукой описано множество специфических нарушений подобного рода. Патологии одного гена бывают аутосомными, или сцепленными с X-хромосомой, рецессивными или доминантными.
Доминантный аутосомный признак
Лишь одна аутосомная аллель гена нужна, чтобы выразить аутосомные доминантные черты; это означает, что происходит поражение гомозиготы и гетерозиготы аномального гена.
В данном случае применимы такие правила:
1. Мужчина и женщина подвержены одинаковому риску появления болезни.
2. У больного человека будет больной родитель.
3. Здоровый ребенок больного родителя не передает черту своему потомку.
4. Здоровый родитель и гетерозиготный больной родитель обладают, в среднем, одинаковым количеством здоровых и больных детей; это означает, что вероятность развития заболевания составляет 50 процентов для каждого потомка.
Аутосомный рецессивный признак
Чтобы выразить аутосомную рецессивную черту необходимо наличие двух копий аномальной аллели. У определенных поколений процент гетерозиготных носителей является высоким по причине эффекта инициатора (то есть была начата группа несколькими людьми, из которых один был носителем) либо вследствие того, что носители обладают селективным преимуществом (к примеру, гетерозиготность в случае серповидно-клеточной болезни служит защитой от малярии).
В данном случае применимы такие правила наследования:
Когда у здоровых родителей был рожден больной ребенок, оба родителя являются гетерозиготными носителями и, в среднем, один из 4х их потомков будет болен, один из 2х гетерозиготный, а один из 4х– здоровым.
В среднем, половина детей больного человека, а также один гетерозиготный носитель подвержены заражению, в треть является гетерозиготными носителями.
Все дети двух больных родителей будут больны.
Женщины и мужчины в одинаковой степени подвержены риску заражения.
Гетерозиготные носители фенотипически нормальны, однако выступают проводниками черты. Когда черта порождена дефектом специфического белка (к примеру, энзимы), обычно гетерозиготный человек имеет ограниченное количество этого белка. Когда нарушение известно, с помощью генетических молекулярных приемов возможно проведение идентификации гетерозиготных носителей.
Родственники скорее прочих унаследуют такую же мутантную аллель, поэтому браки между близкими родственниками сильно увеличивают вероятность рождения больных детей. У пары брат-сестра или родитель-ребенок вероятность родить нездорового ребенка возрастает за счет наличия 50 процентов одинаковых генов.
Невынашивание беременности и женское бесплодие – генетические аспекты

Патологии женской репродуктивной системы в целом, и невынашивание беременности, как важная часть этой проблемы тесно связаны с генетикой человека. Так, наиболее значимые звенья сложного и многосоставного патогенеза акушерских патологий имеют своей причиной мутации генов, обеспечивающих самые разнообразные процессы, участвующие в возникновении и нормальном протекании беременности. Рассмотрим несколько основных акушерских патологий с генетической точки зрения.
Тромбофилия
Тромбофилия – термин, обозначающий предрасположенность к венозному и случайному артериальному тромбоэмболизму. Патологическая способность к тромбообразованию может быть результатом врожденного или приобретенного нарушения свертывания крови. Наиболее распространена активация резистентности протеина С, которая вызвана мутацией фактора 5 Лейден (5)
Тромбофилия играет важную роль в патогенезе развития:
Тромбофилия является ключевой проблемой в процессе развития акушерских осложнений во время беременности:
Венозные тромбозы занимают третье место по распространённости среди сердечно-сосудистых заболеваний. Частота венозных тромбозов в общей популяции, согласно мировым данным, составляет 1-2 случая на 1000 населения ежегодно.
Риск тромбозов значительно увеличивается при наличии таких провоцирующих факторов, как хирургические операции, приём гормональных контрацептивов, различные заболевания (диабет, онкология, ожирение), а также беременность и роды.
Планирование беременности.
Своевременно выявленная тромбофилия – не приговор, а прямая инструкция к действию, ведь своевременно, еще до наступления беременности, терапия позволяет минимизировать, а часто и полностью исключить патологии беременности, вызванные тромбофилией.
Выбор способа контрацепции.
При приеме эстрогенсодержащих контрацептивов свертываемость крови повышается, что в случае тромбофилии становится просто опасным для здоровья и жизни.
Показания к назначению исследования на наследственную тромбофилию:
Факторы риска тромбофилий:
Основные блоки генов, участвующих в развитии тромбофилии:
К первичным (генетически обусловленным) тромбофилиям относят (10):
Частота различных тромбофилий (10):
Основные мутации, приводящие к наследственной тромбофилии:
Коагуляционный фактор 5
Лейденская мутация; G1691A; Arg506Gln
Замедление деградации фактора 5 активированным протеином
Коагуляционный фактор 2 (протромбин)
Усиление синтеза протромбина аллеля А, смещение равновесия в системе гемостаза в сторону усиления свертывания крови
Коагуляционный фактор 2 (протромбин)
Повышение экспрессии гена фибриногена, увеличение выработки фибрина, повышенное тромбообразование
Снижение интенсивности синтеза фибриногена
Образование термолабильной формы фермента со сниженной активностью приводит к нарушению метилирования гомоцистеина и повышению его уровня в крови. Гипергомоцистеинемия приводит к поврежению эндотелия сосудов.
Мутация приводит к термолабильности MTHFR ТОЛЬКО при СОВМEСТНОМ носительстве с мутацией 677T
Усиление плотности рецепторов к коллагену на поверхности тромбоцитов, усиление адгезии тромбоцитов к эндотелию
Мутация влияет на агрегационный свойства тромбоцитов
Ингибитор активатора плазминогена 1-го типа
Аутосомно-доминантная тромбофилия вследствие дефицита протеина С
Аутосомно-доминантная тромбофилия вследствие дефицита протеина С
Аутосомно-доминантная тромбофилия вследствие дефицита протеина S
Аутосомно-доминантная тромбофилия вследствие дефицита протеина S
Тромбофилии в акушерской практике
В настоящее время тромбофилии являются причиной более чем 45 % случаев невынашивания беременности, 80 % преэклампсий, нередко приводя к преждевременной отслойке нормально расположенной плаценты (ПОНРП), синдрому задержки роста плода (СЗРП) с его антенатальной гибелью, а также к последовым и послеродовым кровотечениям (2, 4)
Связь тромбофилии с патологиями беременности:
F5 Лейденская мутация (G1691A)
F2 Коагуляционный фактор 2 / Протромбин (G20210A)
Риск развития тромбозов у носителей аллеля А возрастает в 2-5 раз. У беременных при наличии этой мутации значительно возрастает риск не только венозных, но и артериальных тромбозов, что может приводить к развитию инсультов и ИБС.
FGB Фириноген (G-455A)
Мутация является фактором риска периферического и коронарного тромбоза, ассоциирована со степенью атеросклеротического поражения сосудов. При беременности возрастает риск тромбозов, невынашивания беременности, тромбоэмболических осложнений в родах и послеродовом периоде.
F7 Коагуляционный фактор 7 (G10976A)
SERPINE 1 Ингибитор активатора плазминогена 1-го типа (5G>4G)
Мутации в генах тромбоцитарных факторов
MTHFR 5,10-метилентетрагидрофолатредуктаза (C677T)
Дефекты генов фолатного цикла и беременность
Определённую роль в невынашивании беременности могут играть гены, принимающие участие в метаболизме фолиевой кислоты и витамина В12.
Фолиевая кислота (витамин В9) отвечает за перенос метильных групп (СН3-) при химических реакциях в организме – это реакции, в ходе которых синтезируются ДНК, РНК и ряд аминокислот (глицин, метионин). Поэтому для беременных и для плода так важно не иметь дефицита как самой фолиевой кислоты, так и ферментов, участвующих в ее метаболизме.
Высокие концентрации активной формы фолиевой кислоты необходимы для превращения гомоцистеина в метионин. Основными генами, продукты которых контролируют превращение фолиевой кислоты в метаболически активные формы и регулируют обмен гомоцистеина, являют ся MTHFR (метилентетрагидрофолатредуктаза), MTR (метионинредуктаза) и MTRR (метионинсинтетазредуктаза). Снижение активности этих ферментов – одна из важных причин накопления гомоцистеина в организме.
Схема метаболизма фолиевой кислоты
Причины токсического действия избытка гомоцистеина на организм женщины и плода следующие (6):
Полиморфизм: названия и [синонимы]
C677T; Ala222Val; A222V
Мутантный вариант этого полиморфизма 677Т приводит к образованию термолабильного фермента и повышению уровня гомоцистеина в крови
A1298C; Glu429Ala; E429A
Повышенная частота генотипов А/С и С/С для этого полиморфизма была зарегистрирована у женщин с повторными выкидышами раннего срока.
Показано увеличение частоты встречаемости аллеля 66А гена MTRR и аллеля 2756G гена MTR у женщин с преждевременными родами в анамнезе (после 22-й недели беременности).
Полиморфизм гена MTRR A66C связан с синдромом Дауна и дефектами спинного мозга.
Нарушение имплантации эмбриона (гены TP53, LIF и другие)
На настоящий момент доказано, что экспрессия белка LIF является круциальным фактором для формирования так называемого «окна имплантации».
В эндометрии женщин с бесплодием снижена экспрессия LIF, а также было показано, что при бесплодии неясного генеза значительно снижается экспрессия LIF в среднюю секреторную фазу эндометрия.
Уровень LIF регулируется белком р53, соответственно, мутации генов сигнального пути гена TH53 опосредованно влияют и на экспрессию LIF в эндометрии. Это мутации генов TP53, MDM2, MDM4, USP7 и некоторых других (D.d’Avila Paskulin et al., 2012)
Нарушения имплантации эмбриона (генная сеть генов LIF и TP53)
Эстрогеновый рецептор 1
XbaI Polymorphism; A-351G; IVS1-351A>G
Совместное регулирование как р53 так и эстрогенами требуется для обеспечения экспрессии LIF на уровне, достаточном для имплантации бластоцисты. Учитывая, что функция эстрогенового рецептора при варианте гена rs9340799-G в гомозиготном состоянии (генотип G/G) значительно снижена, следует ожидать уменьшения количества LIF и, как следствие, нарушения имплантации бластоцисты.
Эстрогеновый рецептор 1
Совместное регулирование как р53 так и эстрогенами требуется для обеспечения экспрессии LIF на уровне, достаточном для имплантации бластоцисты. Учитывая, что функция эстрогенового рецептора при варианте гена rs2234693-C в гомозиготном состоянии (генотип C/C) значительно снижена, следует ожидать уменьшения количества LIF и, как следствие, нарушения имплантации бластоцисты.
5-гидрокситриптаминовый (серотониновый) рецептор 1A
У гомозиготных носителей варианта rs6295-C гена 5-HT1A (генотип C/C) обнаруживаются значительно более низкие показатели имплантации. Для них также свойственны значительно более высокие биохимические показатели потери беременности. PMID: 23499153
Фактор, ингибирующий лейкемию
Val64Met; Val86Met; 3400 G/A; G3400A; Arg26His
Фактор, ингибирующий лейкемию
Выявлена ассоциация варианта C полиморфизма rs929271 в 3 ‘UTR гена LIF с нарушением имплантации бластоцисты. Встречаемость C аллеля много выше у женщин с идиопатическим бесплодием в возрасте до 35 лет, но не старше.
MDM2 протоонкоген, E3 убиквитин-протеин-лигаза
Вариант rs2279744-G резко увеличивает экспрессию кодируемого MDM2 белка. Повышенное количество MDM2, в свою очередь, ингибирует p53, что снижает уровень LIF и вероятность имплантации.
MDM4, регулятор p53
Присутствие аллеля Т в полиморфизме rs1563828 гена MDM4 увеличивает экспрессию кодируемого этим геном белка-супрессора р53. Ингибирование р53, в свою очередь, приводит к снижению количества LIF и вероятности имплантации.
Простагландин-эндопероксид синтаза (простагландин G/H синтаза и циклооксигеназа)
Нейротрансмиттерный переносчик серотонина
L/S; Long/Short; 44-bp Ins/Del
У гомозиготных носителей (генотип L/L) обнаруживаются значительно более низкие показатели имплантации, более низкие клинические показатели беременности (PR) и более высокие биохимические показатели потери беременности (BPL).
Опухолевый протеин р53
Arg72Pro; Ex4+119C>G; 12139G>C; R72P
Процент женщин-носительниц аллеля Pro72 был существенно выше в выборке перенесших ЭКО в связи с повышенным числом случаев нарушений имплантации. Аллель Pro72 служит фактором риска развития недостаточности имплантации.
Убиквитин-специфическая пептидаза 7, ассоциированная с вирусом герпеса
У пациентов, несущих аллель Hausp A (генотип A/A), наблюдается значительное нарушение функции гена USP7, снижающее в итоге активность p53. Потеря активности p53 влечёт за собой снижение экспрессии гена LIF, что уменьшает количество соответствующего белка, играющего ключевую роль в процессе имплантации бластоцисты
Преэклампсия
Преэклампсия – возникающий при беременности синдром полиорганной недостаточности, в основе которого лежит увеличение проницаемости сосудстой стенки и других мембран исвязанные с этим волемические и гемодинамические нарушения.
На генетическую предрасположенность к преэклампсии влияет целый ряд генетических полиморфизмов. В настоящее время исследованы свыше 20 генов предрасположенности к гестозу, однако данные постоянно перепроверяются и пополняются.
Четко показана связь с гестозом для следующих групп генов:
Генетическая предрасположенность преэклампсии (эндотелиальной дисфункции, гестозу)
Полиморфизм: названия и [синонимы]
Эстрогеновый рецептор 1
XbaI Polymorphism; A-351G; [IVS1-351A>G]
При одновременном наличии Т/Т генотипа полиморфизма Pvull, риск преэклампсии возрастает в 3,26 раза даже при отсуствии таких факторов риска, как повышенный индекс массы тела, курение, беременность.
Эстрогеновый рецептор 1
PvuII Polymorphism; T-397C; [-397T>C]
При одновременном наличии A/A генотипа полиморфизма Xball, риск преэклампсии возрастает в 3,26 раза даже при отсуствии таких факторов риска, как повышенный индекс массы тела, курение, беременность.
Met235Thr; M235T; Met268Thr; M268T
У носительниц аллеля 235Thr повышена концентрация ангитензиногена в крови; риск развития преэклампсии в 2 раза выше, чем у носительниц «дикого» аллеля Met235.
Ins/Del, Intron 16; 289bp Alu-Ins/Del
У носительниц аллеля D упровень АФП повышен; риск развития преэклампсии в 2 раза выше, чем у носительниц «дикого» аллеля I.
Ангиотензин-превращающий фермент (Дипептидилкарбоксипептидаза 1)
Ins/Del, Intron 16; 289bp Alu-Ins/Del
Мутации гена АСЕ являются одной из причин эндотелиальной дисфункции. У носительниц генотипа D/D уровень АПФ повышен; риск развития преэклампсии и тромбоэмболий в 2 раза выше, чем у носительниц «дикого» генотипа I/I.
Синтаза оксида азота 3
Glu298Asp; E298D; [G894T; 894G>T]
Показано, что у гомозигот по минорному аллелю Asp/Asp риск развития преэклампсии выше по сравнению со среднепопуляционным в 2,45 раза. Носительницы аллеля Asp (генотипы Asp/Asp и Glu/Asp) встречаются чаще в группах беременных, страдающих гипертонией и отслойкой плаценты, по сравнению с группой женщин без данных осложнений беременности. Наличие аллеля Asp является фактором риска развития гипертензии при беременности.
T-786C; [-786 T/C; IVS1-762C>T]
Замена С на Т в регуляторной области гена ведет к снижению экспрессии гена, которая ведет к повышению тонуса артерий. Это является фактором риска спазма коронарных сосудов, в сочетании с присутствием аллеля 298Asp значительно повышает риск развития преэклампсии.
Активатор ингибитора плазминогена, тип 1 (PAI1)
4G/5G; PAI1: 4G/5G; Ins/Del G; [-675 4G/5G; Ins/Del(G)]
Полиморфизм 4G/5G является полиморфизмом инсерции/делеции нуклеотида G в промоторной области гена SERPINE1. Отсутствие одного нуклеотида в области полиморфизма получило название 4G варианта, который связан с высокой транскрипционной активностью данного гена и увеличением концентрации его продукта, что, в свою очередь, негативно воздействует на фибринолиз, повышает риск сосудистых осложнений и тромбозов. Наличие варианта 4G в гомозиготном состоянии (генотип 4G/4G) связано со значительным риском развития поликистоза яичников, а также со значительным риском возникновения осложений беременности, таких как ранние эмбриональные и преэмбриональные потери, гестозы и неудачи ЭКО. Указанные риски увеличиваются при сочетании с носительством варианта 34Leu гена полипептида A1 фактора коагуляции XIII (F13A1).
Фактор коагуляции XIII, полипептид A1
Val34Leu; Val35Leu; 9G>T
Данная мутация приводит к дефициту фактора свертывания 13, вызванной отсутствием субъединица А белка. Этот дефект приводит к склонности к кровотечениям, плохому заживлению ран и привычным выкидышам.
Интерлейкин 6 (интерферон, бета 2)
Гиперандрогения и синдром поликистозных яичников (СПКЯ)
Согласно консенсусу специального Международного симпозиума объединенной рабочей группы ESHRE/ASRM (Европейского общества репродукции и эмбриологии человека и Американского общества репродуктивной медицины). Симпозиум состоялся 1–3 мая 2003 г. в Роттердаме (Нидерланды), на нем были приняты следующие диагностические критерии постановки диагноза наличие хотя бы двух из трех утвержденных критериев позволяет после исключения иных состояний верифицировать диагноз СПКЯ. Таким образом, с одной стороны, в плане обследования СПКЯ остается синдромом (комплексом симптомов), идентификация которого невозможна и недопустима на основании изолированного наличия любого единственного диагностического критерия (одиночно от остальных двух). С другой стороны, простой анализ попарного сочетания современных критериев позволяет сделать принципиальный вывод о необходимости расширенного толкования термина СПКЯ. Это связано с дополнительным включением в его дефиницию новых клинических форм, а именно: в отсутствие другой гиперандрогенной патологии диагноз СПКЯ допустимо выставлять не только при классическим течении (полной триаде признаков), но и при наличии одного из трех неполных (неклассических) клинико-инструментальных дуэтов.
Диагностические критерии СПКЯ (по консенсусу Симпозиума рабочей группы ESHRE/ASRM):
Основные мутации, приводящие к развитию СПКЯ
(CAG)n repeat; (3bp)n, Short/Long (S/L)
При уменьшенном количестве (CAG)-повторов у женщин возрастает риск развития рака груди, эндометрия, синдрома поликистозных яичников и гиперандрогении (является фактором риска развития инсулинорезистетности, но лишь при наличии других факторов риска).
Мутации в гене SRD5A1 повышают уровень активности фермента, что приводит к увеличению синтеза дигидротестостерона. Дигидротестостерон обладает более выраженным андрогенным эффектом, чем тестостерон.
Фермент преобразует прегненолон и прогестерон в дегидроэпиандростерон (DHEA) и андростендион. Мутации в этом гене связаны с псевдогермафродитизмом и гиперплазией коры надпочечников.
Цитохром P450, семейство 11, подсемейство А, полипептид 1 (Фермент, расщепляющий боковую цепь холестерина)
При увеличении числа (TTTTA)n повторов (вариант L, где n>16) в гене CYP11A1 повышается продукция андрогенов, что является фактором риска развития СПКЯ.
Цитохром P450, семейство 21, подсемейство A, полипептид 2
Мутация ведет к развитию неклассической (постпубертатной) формы врожденной дисфункции коры надпочечников. При этой форме заболевания первые признаки появляются только во время полового созревания. Высок риск бесплодия и невынашивания беременности.
Показано, что риск развития ановуляции у носительниц аллеля III гена INS повышен по сравнению с носительницами генотипа I/I. Это связано с тем, что гиперинсулинемия/резистентность к инсулину опосредует снижение чувствительности гипофиза к гонадотропин-релизинг гормону, что способствует ановуляции.
Рецептор гамма, активируемый пролифератором пероксисом
Pro12Ala; P12A (протективный)
У носителей мутантного аллеля 12Ala повышена чувствительность к инсулину. Это является протективным фактором в отношении риска развития диабета 2 типа и СПКЯ. Однако, протективный эффект нивелируется при ожирении и повышении индекса массы тела.
Показания к назначению исследования на СПКЯ (8):


