Низкое расположение миндалин мозжечка чем опасно
Информация о работе и расписание
Госпитальная высококвалифицированная медицинская помощь
Услуги центра по восстановительной медицине
Восстановление после спортивных травм
Современная диагностика – шанс предупредить болезнь
Он-лайн консультации для врачей по сложным практическим случаям
Трудоустройство в ФГАУ ЛРЦ
Стандарты и порядки оказания медицинской помощи
Проведение этической экспертизы клинических исследований, медицинских испытаний
Статьи и презентации
Мальформация Киари III типа заключается в смещении мозжечка и части ствола мозга с мозговыми оболочками в менингоцеле, расположенное в шейно-затылочной области.
Мальформации Арнольда–Киари II и III типов могут сопутствовать признаки дисплазии нервной системы: полимикрогирия, гетеротопия коры, гипоплазия подкорковых узлов, дисгенезия мозолистого тела, патология прозрачной перегородки, утолщение интерталамического соединения, beaking tectum (клювовидный tectum), часто отмечают наличие перегиба сильвиевого водопровода (55%), кисты отверстия Мажанди, гипоплазия серпа и намета мозжечка, hemivertebrae, низкое расположение каудального отдела спинного мозга на уровне LIV–V позвонков и ниже.
Этиология заболевания в настоящее время не ясна. Имеются данные, свидетельствующие о роли генетического фактора в этиологии этого синдрома. Эктопия миндалин мозжечка в затылочное отверстие была обнаружена у трех монозиготных близнецов. После первого описания мальформации Cleland в 1883 г. появилось несколько теорий. Теория, подтверждаемая исследованиями Misao Nishikawa и соавторов, заключается в том, что из-за парааксиальной дисплазии мезодермального листка или первичного повреждения структур соответствующего сомита формируется ненормально маленькая задняя черепная ямка, структуры заднего мозга, заполнив объем задней черепной ямки и продолжая расти, опускаются в затылочный канал. Сочетание Аномалии Киари II типа с менингомиелоцеле связано с тем, что степень парааксиальной дисплазии мезодермального листка при АК – II типа более выражена, чем при АК – I типа и отмечается не только на уровне формирования затылочной кости, но и по оси тела на уровне формирования ряда позвонков, что проявляется в spina bifida, а также в аномалиях ряда других костных структур и костной системы в целом.
Клинические проявления АК – I типа проявляются чаще всего в юношеском либо в зрелом возрасте. Эти проявления укладываются в такие неврологические синдромы, как церебеллобульбарный, ликворогипертензионный, сирингомиелический, синдромы повреждения черепных нервов. Ликворогипертензионный синдром проявляется головной болью, обычно субокципитальной, и болью в шее, усиливающейся при кашле, чихании и напряжении, застойными дисками зрительных нервов. Стволовые нарушения и расстройства функций черепных нервов проявляются в виде неустойчивых осциллопсий, тригеминальной дизестезии, снижения слуха, шума в ушах, головокружения, дисфагии, остановки дыхания во время сна, периодических обмороков (часто связанных с кашлем), нарушения контроля над ЧСС, АД при переходе из горизонтального положения в вертикальное, могут наблюдаться атрофия половины языка, паралич голосовых связок, стридор, спастический или комбинированный (больше в верхних конечностях) тетрапарез.
Мозжечковые расстройства — нистагм, дизартрия, атаксия. Симптомы, связанные с сирингомиелическими кистами — онемение, расстройство чувствительности, обычно по диссоциированному типу, а также нейроартропатия, нарушение функций тазовых органов, отсутствие брюшных рефлексов, мышечная гипотрофия. При этом ряд авторов отмечают несоответствие между локализацией, протяженностью кисты, кистозным индексом (отношение переднезаднего размера кисты к таковому размеру поперечника спинного мозга на уровне кисты), с одной стороны, и зоной гипестезии, распространенностью сегментарных расстройств поверхностной чувствительности, выраженностью мышечной гипотрофии и степенью пареза — с другой. АК II типа манифестирует у новорожденных и в раннем детском возрасте такими симптомами, как апноэ, стридор, билатеральный парез голосовых связок, нейрогенная дисфагия с назальной регургитацией, цианоз во время кормления, нистагм, гипотония, слабость, спастика в верхних конечностях, что может прогрессировать вплоть до тетраплегии. Мальформация Киари III типа встречается редко, клинические проявления ее такие же, как при АК II.
Стандартное рентгенологическое исследование может выявить лишь косвенные признаки мальформации АК, компьютерная томография также не дает четкой визуализации мягкотканных структур. Широкое внедрение МРТ в клиническую практику позволило решить большинство проблем, связанных с диагностикой аномалии Киари. Этому способствовали хорошая визуализация структур задней черепной ямки, краниовертебрального перехода, спинного мозга, отсутствие артефактов от костных структур.
Ориентиры задней черепной ямки, используемые в диагностике АК. d + e = длина ската; S = сфеноокципитальный синхондроз; d = длина основания сфеноидальной площадки от спинки турецкого седла и сфеноокципитального синхондроза до ската; e = длина между синхондрозом и basion; b = длина ствола мозга между плоскостью соединения среднего мозга и моста и медулло-цервикальным соединением; a = угол намета мозжечка по отношению к линии Твайнинга (Twining’s line); c = длина полушария мозжечка; DS = верхушка спинки турецкого седла; IOP = внутреннее возвышение затылочной кости; OP = opisthion; B = basion; TW = линия Твайнинга; McR (B to OP) = линия МакРи (McRae’s line). (заимствовано из Dimensions of the posterior fossa in patients symptomatic for Chiari I malformation but without cerebellar tonsillar descent, Raymond F Sekula и соавт.).
Низкое расположение миндалин мозжечка чем опасно
В последнем десятилетии XIX века немецкий патологоанатом Киари описал четыре врожденные аномалии мозжечка и ствола мозга, как проявления нарастающей тяжести единого патогенетического механизма, связанного с давлением сзади на задний мозг при врожденной гидроцефалии.
Основной анатомической особенностью аномалии Киари I типа является разная степень каудального смещения миндалин мозжечка в шейный канал. Аномалия Киари 2 типа характеризуется более тяжелой мозжечковой грыжей, которая, кроме миндалин мозжечка, также включает в себя нижний червь и четвертый желудочек; она, как правило, связана с миеломенингоцеле. Аномалия Киари 3 типа включает в себя признаки высокого менингоэнцефалоцеле с грыжей заднего мозга. Наконец, Киари 4 типа характеризуется тяжелой гипоплазией мозжечка без каудального смещения.
После многих лет и большого количества теоретических, клинических и нейровизуализационных исследований, понимание и осознание этого состояния вышло на новый уровень. В настоящее время считается, что эти четыре порока развития являются отдельными состояниями, с различным патогенезом, клиническим проявлением, и прогнозом, а не разными прогрессирующими стадиями одного заболевания. С анатомической точки зрения аномалии 1-3 типа являются грыжами заднего мозга различной степени, с возможным вторичным образованием полости в спинном мозге, в то время как аномалия 4 типа характеризуется гипоплазией мозжечка. Гипоплазия задней черепной ямки является частой находкой при первых трех аномалиях, но никогда не встречается при четвертой.
У каждой формы свои патогномоничные клинические проявления и по этим причинам различные аномалии Киари должны рассматриваться в разных разделах.
Мальформация Киари 1 типа. Аномалия Киари 1 типа характеризуется неправильным положением (и формой) миндалин мозжечка, которые опускаются из полости черепа ниже большого затылочного отверстия в шейный канал. Это анатомическое нарушение приводит к окклюзии субарахноидального пространства на уровне большого затылочного отверстия с последующим нарушением ликвороциркуляции в головном и спинном мозге; фактически, помимо изменений в задней черепной ямке, повышение ликворного давления на этом уровне приводит к сирингомиелии. Патогенетические механизмы развития аномалии детально изучены и обсуждены в литературе. Различные механизмы патогенеза можно схематично разделить на три категории:
— Гидродинамический (на основе градиента давления между ликворными пространствами головного и спинного мозга).
— Механический (с блоком ликвороциркуляции на уровне большого затылочного отверстия как причины аномалии Киари 1 типа).
— Нарушение развития (с интерпретацией аномалии задней черепной ямки как местного проявления более общего нарушения развития).
Хотя аномалия Киари 1 типа обычно становится симптоматичной в раннем подростковом периоде, в последнее время она обнаруживается с одинаковой частотой как у детей, так и у взрослых.
а) Клиническая картина аномалии Киари I типа. Наиболее частой жалобой у этой группы пациентов является боль в затылке или в шее, усиливающаяся при чихании, кашле или при пробе Вальсальвы. Другими болезненными проявлениями являются боль в плече, спине или боли в конечностях без корешкового распределения. Наиболее частым симптомом являются признаки моторного или сенсорного дефицита в конечностях (> 70%), которые являются проявлением наличия полости в спинном мозге (сирингомиелии). Атаксия тела и конечностей в качестве проявление нарушений в мозжечке является вторым наиболее распространенным симптомом (в 30-40%). Реже (в 15-25%) наблюдается неуклюжесть, нистагм, диплопия, дисфагия и дизартрия как проявление дефицита черепных нервов. Апноэ при заикании наблюдается в 10% случаев, в большинстве случаев проявляясь у младенцев или маленьких детей. Своеобразным проявлением аномалии Киари 1 типа у детей и подростков является прогрессирующий сколиоз (в 30%).
б) Лучевая диагностика. МРТ — лучший способ для диагностики аномалии Киари 1 типа. Важными критериями для этого порока являются: пролабирование одного или обоих миндалин мозжечка ниже большого затылочного отверстия (> 5 мм); возможны шейно-мозговая деформация, отсутствие супратенториальных нарушений (за исключением отдельных случаев небольшого расширения желудочков и обычная локализация четвертого желудочка. Аномалия Киари 1 типа может быть связана с такими костными аномалиями, как маленькая задняя черепная ямка, платибазия, атланто-затылочная ассимиляция, базилярная импрессия, срастание шейных позвонков (аномалия Клиппеля-Фейля). Гидро/сирингомиелия встречается в 50-60% случаев. Она может быть ограничена одной или двумя областями, а может распространяться на всю длину спинного мозга. Обычно полость образуется на уровне С1.
МРТ является полезным дополнением к диагностическим мероприятиям, так как позволяет определить степень сдавления ствола на уровне затылочного отверстия и характеристики ликвородинамики; исследование, выполненное в послеоперационном периоде, позволяет получить некоторое представление об адекватности хирургической декомпрессии.
в) Лечение аномалии Киари I типа. Целью хирургического лечения является декомпрессия задней черепной ямки для восстановления циркуляции спинномозговой жидкости в базальных цистернах и устранение сдавления нервных структур на уровне краниоцервикального перехода. Хирургическое лечение при аномалии Киари 1 типа обычно определяет четко определенный протокол, который включает субокципитальную краниотомию, С1 ламинэктомию, лизис арахноидальных сращений, резекцию миндалин мозжечка (как традиционную тонзилэктомию, так и субпиальную коагуляцию) и расширенную дуропластику. В случае сопутствующей вентральной компрессии (как, например, при платибазии, С1 ассимиляции и т.д.), она должна быть устранена до проведения дорсальной декомпрессии. В последнее время появляются сообщения, которые указывают на возможность достижения сопоставимых результатов при использовании простой декомпрессии, без расширяющейся дуропластики (или с отслоением наружного слоя твердой мозговой оболочки). Интраоперационная ультразвуковая диагностика может быть полезна в этом отношении, демонстрируя синхронизацию движения миндалин с дыханием и сердцебиением, а также наличие адекватного движения ликвора из четвертого желудочка.
Дальнейшие хирургические возможности представлены процедурами, направленными на лечение гидро/сирингомиелии (закупорка задвижки, размещение сиринго-субарахноидального или сиринго-плеврального шунта, и стентирование четвертого желудочка). При сравнении двух основных крупных хирургических вмешательств при лечении аномалии Киари 1 типа с сирингомелией — субокципитальной декомпрессии и установки сиринго-субарахноидального шунта—Hida et al. обнаружили уменьшение размера сирингомиелитической полости у 94% пациентов, перенесших декомпрессию, и у 100% перенесших сирингосубарахноидальное шунтирование. Что касается прогноза, то максимальные преимущества декомпрессии проявляются у пациентов с симптомами, связанными с пароксизмальной внутричерепной гипертензией. Кроме того, пациенты с мозжечковой симптоматикой и синдромом большого затылочного отверстия имеют больше шансов на выздоровление, чем пациенты с центральным синдромом спинного мозга.
При работе с пациентами детского возраста главной проблемой являются показания к операции. Фактически, во многих случаях диагноз аномалии Киари ставится случайно, после МРТ для диагностики причины неспецифических клинических проявлений, таких как головные боли, умственная отсталость, эпилепсия. Большинство авторов не согласно с превентивным хирургическим лечением, считая, что показания к нему должны основываться на наличии симптомов и клинических проявлений, однозначно относящихся к аномалии Киари, а не данными нейровизуализации.
Что касается хирургической техники, некоторые авторы используют те же оперативные приемы, что и для взрослых, т. е. субокципитальную декомпрессию, С1 ламинэктомию, пластику ТМО и резекцию миндалин мозжечка в 40-60% случаев. При послеоперационной МРТ отмечается 100% улучшение симптомов, а также более чем 90% улучшение признаков, связанных с сирингомиелией и уменьшение сирингомиелитических кист в 80% случаев. В последние годы некоторые авторы критикуют этот «классический» подход как слишком тяжелый (а именно интрадуральные манипуляции), особенно в случаях умеренного опущения миндалин. В действительности, по некоторым сообщениям существуют указания на возможность достижения сопоставимых результатов менее инвазивным методом, то есть выполнением субокципитальной декомпрессии (и С1 ламинэктомия) с или без расслаивания внешнего слоя твердой мозговой оболочки. Улучшение или разрешение клинических проявлений описывается у более чем 90% детей, а исчезновение сирингомиелической полости — в 80% случаев. Удаление миндалин мозжечка с вскрытием ТМО или дуропластикой должно использоваться в случаях неэффективности минимально инвазивного лечения.
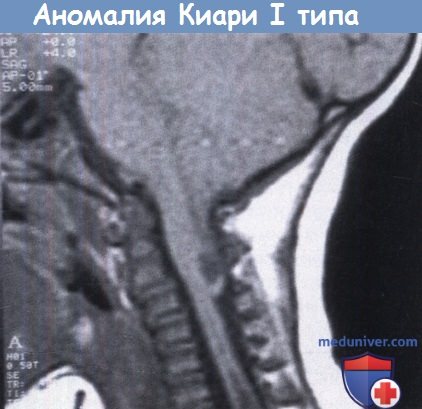 Аномалия Киари 1 типа.
Аномалия Киари 1 типа.
Сагиттальный срез в Т1-взвешенном режиме МРТ.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Арнольда — Киар
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Что для организма означает низкое расположение миндалин мозжечка?
Опущение миндалин мозжечка в большое затылочное отверстие или кагал спинного мозга называют дистопией. А иногда эта патология называется аномалией Киари. Как правило, такое заболевание не влечет за собой значительных расстройств или явных симптомов и не имеет причин для беспокойства больного. Часто эта патология проявляется после достижения 30 – 40-летнего возраста. Обнаруживается обычно при обследованиях по другим причинам. Потому об этом заболевании следует знать, чтобы оно не оказалось неожиданностью для больного.
Для того, чтобы понять, что такое низкое расположение миндалин мозжечка, необходимо четко знать клинические симптомы и как обнаруживается патология. А поскольку это состояние сопровождается частыми головными болями, нужно сначала установить их причину, а потом уже заниматься лечением. Такое заболевание выявляется при МРТ.
Причины появления
Дистопия мозжечка, как правило, является врожденной патологией. Она возникает при смещении какого-либо органа в эмбриональный период. Вторичной она бывает лишь при проведении частых пункций или при люмбальных травмах. Иных причин появления данного заболевания не выявлено.
Миндалины мозжечка очень сходны с теми, которые находятся в гортани. В нормальном положении они располагаются выше БЗО черепа. А отклонения в их развитии и положении могут повлечь за собой не только дистопию. Чаще всего встречается опущение миндалин мозжечка ниже уровня черепа.
До сих пор заболевание Киари является патологией, о причинах возникновения которого неврологи не пришли к одному мнению. Некоторые придерживаются мнения, что эта аномалия возникает при уменьшении габаритов ямки позади черепного выходного отверстия к спинному мозговому каналу. Это нередко приводит к таким последствиям в процессе роста тканей, которые расположены в коробке. Они выходят в затылочный выходной канал. Иные специалисты полагают, что заболевание начинает развиваться по причине увеличения объемов мозговых тканей головы. В этом случае мозг начинает выталкивать через заднюю черепную ямку в затылочное черепное отверстие мозжечок и его миндалины.
Вызывает такое прогрессирование выраженной аномалии и ее переход в «клинику», как гидроцефалия. При этом увеличиваются общие размеры мозга, особенно мозжечковые ткани. Патологию Киари вместе с недоразвитым связочным аппаратом мозга сопровождает дисплазия тканей кости. Потому любая черепно-мозговая травма нередко приводит к усилению снижения уровня нахождения миндалин и мозжечка.
Типы аномалий
Есть такие виды аномальных отклонений – дистопия и аномалия Киари.
В свою очередь, заболевание Киари разделяют на четыре различных типа:
II и III типы часто проявляются в сочетании с явлениями дисплазии нервной системы, например, с гетеротопией мозговых тканей коры, кистами отверстия и пр.
Симптомы
Самой распространенной среди аномалий является патология первого вида. При ней нередко возможно проявление ликворно-гипертензионного синдрома, а также церебелло-бульбарное и сирингомиелическое явления, нарушения работы нервных окончаний внутри черепа.
Ликворно-гипертензионный синдром – это боли в затылке и шейных мышцах, которые усиливаются во время чихания, кашля или напряжения шейных мышечных тканей. Часто боли сопровождаются рвотой, не связанной с приемами еды. Многие симптомы патологии проявляются в зависимости от того, в каком положении располагаются миндалины мозжечка относительно отверстия, которое находится в затылочной ямке черепа. Наблюдается также:
Аномалия II и III типов имеет похожие симптомы, заметные уже с первых мгновений после рождения малыша. Второй тип сопровождает шумное дыхание, а также неожиданные приступы остановки дыхания, нейропарез гортанных тканей. Наблюдаются также отклонения процесса глотания.
Признаки дистопии редко бывают очевидными. Но все же возможны неврологические проявления:
Если опущение миндалин сильное, иногда наблюдается расширение канала, связывающего головной и спинной мозг, а также образовываются полости вокруг канала.
Диагностические методы
Основным современным методом диагностирования дистопии является МРТ. В этом случае ни КТ, ни рентгеновские исследования не дают полной картины патологии.
Для диагностирования синдрома Киари не подойдут никакие стандартные методы типа ЭЭГ, ЭхоЭГ или РЭГ, потому как они не позволяют точно поставить диагноз. Осмотр невролога тоже не определит аномалию. Все эти методы могут показать подозрение только на повышенное давление внутри черепной коробки. Рентгенографию черепа также не стоит делать, поскольку она показывает лишь аномалии костных тканей, которые могут сопровождать патологию. Потому до введения в диагностическую практику томографии диагностировать эту болезнь было проблематично. Современные же способы диагностирования позволяют точно определить патологию.
В случае качественной визуализации костных тканей вертебрального перехода такие методы, как МСКТ или КТ, не дают достаточно точной картины. Единственным достоверным способом диагностирования аномалии Киари на сегодня является только МРТ.
Поскольку проведение исследования этим методом требует неподвижности больного, маленьких детей погружают в искусственный сон с помощью лекарств. Проводится также МРТ спинного мозга. Она направлена на диагностику любых аномальных отклонений в работе нервной системы.
Лечение
Консервативные методы лечения возможны лишь при очень незначительных отклонениях. Все зависит от того, каково состояние больного на момент обращения к врачу. В этом случае лечение направлено на снятие болезненных симптомов нестероидными лекарствами или миорелаксантами. Необходима также коррекция режима.
Единственным эффективным методом лечения при обширных отклонениях является хирургическое вмешательство, которое заключается в расширении ямки черепа и пластики твердых мозговых тканей оболочки.
Показаниями для оперативного лечения являются:
Если аномальное отклонение протекает без каких-то ощутимых признаков, лечения не требуется. В случаях возникновения болезненных ощущений в районе шеи и затылка проводится консервативная терапия, при которой используют анальгетики и асептические лекарственные вещества, а также миорелаксанты.
При сопровождении аномалии Киари нарушениями неврологических функций или когда консервативный курс терапии не дает результатов, назначается хирургическая операция.
Часто при лечебных курсах синдрома Киари используется метод краниовертебральной декомпрессии. Операция подразумевает расширение отверстия затылочной части за счет удаления части костной ткани, отсечения мозжечковых миндалин и части двух позвонков шеи. Благодаря этому, нормализуется оборот цереброспинальной жидкости в мозговых тканях в результате выполнения заплаты из аллотрансплантата или искусственного материала. Иногда синдром Киари лечат с помощью шунтирования, которое позволяет дренировать цереброспинальную жидкость из центрального канала. Посредством хирургической операции можно отвести цереброспинальную жидкость в сосуды органов груди или брюшины.
Прогноз
Аномалия Киари первого типа может протекать бессимптомно всю жизнь. И третий тип патологии почти всегда приводит к смертельному исходу, если не осуществить своевременное лечение. В случае появления неврологических признаков заболевания первого или последнего типов очень важно своевременно проведенное хирургическое лечение, потому как появившийся недостаток неврологических функций будет плохо восстанавливаться, даже если успешно провести манипуляции. По разным данным, результативность хирургической операции отмечается примерно в половине эпизодов.


