Нейрофиброматоз: отражение проблемы в глазах

О клинической картине и типичных нарушениях развития при различных типах нейрофиброматоза — Вера Липковская, Анна Чистопрудова, члены рабочей группы по нейрофиброматозу мрбоои «Союз пациентов и пациентских организаций по редким заболеваниям», ассоциаты европейской организации NF PATIENTS UNITED.
ГЕНЕТИЧЕСКОЕ ОТКЛОНЕНИЕ
Нейрофиброматоз, также известный как болезнь (синдром) Реклингхаузена, представляет собой генетическое заболевание, которое, по разным
данным, наблюдается примерно у 1 из 2500-3500 новорожденных детей. Возникновение болезни может быть обусловлено наследственными факторами,
а в ряде случаев — спонтанной мутацией генов. Гены, ответственные за возникновение нейрофиброматоза, локализованы на длинном плече 17 хромосомы 17q11.2, в которой содержится информация, ответственная за синтез нейрофибромина и других белков. Ген НФ-1 выступает как супрессор опухолевых
процессов, а потому в результате мутации нарушается общий иммунитет к
возникновению опухолей, которые в большинстве случаев являются доброкачественными.
Первые упоминания о данной болезни встречаются в литературе XIX в.,
когда ирландским хирургом Робертом Смитом в отдельную группу были выделены пациенты с опухолями на нервных тканях. Впоследствии заболевание
было описано в 1882 году в монографии немецкого патологоанатома Фридриха фон Реклингхаузена, который описал клиническую картину и патологические основы появления нейрофибром (доброкачественных опухолевых образований). В 1937 году австрийский офтальмолог Карл Лиш описал узелки
Лиша (Lisch nodules) у пациентов с нейрофиброматозом. В 1956 году исследователь Франк Кроуэ и его коллеги впервые определили, что данное заболевание является наследственным и передается потомству в
50% случаев. В 1988 году Национальные институты здравоохранения США (National In itutes of Health Consensus
Development) провели первую конференцию по нейрофиброматозу с целью выработки последовательных критериев его диагностики. По итогам конференции был принят
доклад, в котором выделялось 7 типов нейрофиброматоза.
3 ТИПА ЗАБОЛЕВАНИЯ
В настоящее время в зарубежной практике принято
выделять 3 типа заболевания: нейрофиброматоз I типа, нейрофиброматоз II типа и шванноматоз.
Для удобства нейрофиброматоз I типа и нейрофиброматоз II типа далее также будут сокращенно именоваться НФ1 и НФ2 соответственно.
Диагноз нейрофиброматоз I типа может быть поставлен
при наличии сочетания двух и более симптомов:
появляются в течение первых 2 лет жизни;
Указанные симптомы проявляются у пациентов в различные периоды жизни и практически на всем ее протяжении. Часть из них формируется в течение первых двух
лет жизни, другие к 5-7 годам, и т.д.
Помимо собственно клинической картины заболевания, оно также может стать причиной задержки развития, когнитивных нарушений, проблем психосоциального характера, нарушения координации и т.д.
Выделяют следующие основные группы нарушений в развитии при НФ1, часто являющиеся причиной низкой успеваемости при обучении: моторные навыки; речевые навыки; письмо; память, внимание и мышление;
визуально-пространственное восприятие; проблемы с планированием и организацией; проблемы с поведением и социальным взаимодействием.
Во многих случаях у пациентов с НФ1 наблюдаются расстройства аутистического спектра (РАС) и синдром дефицита внимания и гиперактивности (СДВГ), депрессивные и тревожные состояния.
Нейрофиброматоз — не приговор, и случается, что люди с данным заболеванием живут, совершенно не подозревая о его наличии, пока не проявится какой-либо из клинических симптомов, иногда уже во взрослом возрасте. Вместе с тем, многие пациенты постоянно находятся в зоне риска, поскольку заболевание неизлечимо и может дать осложнение в любой момент. Именно поэтому вопрос своевременной диагностики и доступного медицинского обслуживания стоит особенно остро, так
как пациенты с НФ1 должны постоянно наблюдаться у дерматолога, невролога, ортопеда, офтальмолога, кардиолога, онколога, психолога.
К сожалению, сегодня пациентам с НФ1 в России, особенно, в регионах, не так просто получить качественное медицинское обслуживание в силу того, что не все врачи
имеют большой опыт и знания в области данного заболевания. Понимание особенностей клинической картины НФ1 и дальнейшее изучение и обобщение опыта в этой сфере поможет обеспечить людям, страдающим данным заболеванием, более качественную поддержку и улучшить тем самым качество их жизни.
Нейрофиброматоз II типа встречается реже, примерно у 1 из 50 000 новорожденных. Молекулярно-генетические исследования выявили принципиальные отличия в патогенезе НФ1 и НФ2, которые представляют собой разные заболевания и
требуют дифференцированного клинического подхода. НФ2 формально является аутосомно-доминантным генетическим заболеванием. Возникающие при НФ2 опухоли являются доброкачественными, но биологически более агрессивными, чем при НФ1.
Диагноз нейрофиброматоз II типа может быть поставлен при наличии сочетания двух и более симптомов:
При НФ2 происходит развитие опухолей на восьмом черепно-мозговом нерве и вестибулярных нервах, что зачастую вызывает давление на слуховые нервы и приводит к потере слуха, и данная угроза сохраняется на протяжении всей жизни. Также примерно в подростковом возрасте появляются: звон в ушах, онемение лица,
головокружение, нарушение баланса тела, хронические головные боли. При наличии опухолей спинного мозга возможно онемение других частей тела.
В течение последних нескольких лет зарубежные ученые ведут дискуссию о пересмотре диагностических критериев НФ2. В частности, в очередной раз намерение
по пересмотру озвучивалось его инициаторами в рамках Международной конференции по нейрофиброматозу, прошедшей осенью 2018 года в Париже (Франция). Шванноматоз, также иногда именуемый нейрофиброматоз III типа, представляет собой крайне редкое генетическое заболевание, встречающееся с частотой примерно 1 случай на 1,7 млн человек. Впервые данное заболевание было описано
у пациентов из Японии, у которых наблюдались множественные кожные шванномы, опухоли ЦНС и другие неврологические осложнения, однако в отсутствие типичных для НФ клинических симптомов.
В основном, типичные для НФ2 и шванноматоза доброкачественные опухоли вырастают из т.н. шванновских клеток — глиальных клеток, образующих миелиновую оболочку нервов. В случае, когда шванновские клетки начинают бесконтрольно распространяться, они образуют своего рода капсулу, которая называется шванномой.
Несмотря на то, что сами по себе шванномы являются доброкачественными, их наличие может стать опасным в случаях, когда их разрастание приводит к сдавливанию нервов, возникновению хронических болей, что зачастую обусловливает
необходимость хирургического вмешательства или применения других методов
лечения. В частности, хронические боли у пациентов с шванноматозом являются предметом углубленного изучения в зарубежной науке, поскольку их наличие
крайне негативно сказывается на качестве жизни пациентов.
Нейрофиброматоз диагностические критерии у взрослых что это
а) Синоним. Нейрофиброматоз 1 типа (НФ1) также известен как периферический нейрофиброматоз (НФ) или болезнь фон Реклингхаузена.
б) Эпидемиология. Наиболее распространенная форма нейрофиброматоза (НФ) составляет около 96% случаев с заболеваемостью I на 4000 живорожденных. Гендерное и расовое преобладание не описано.
в) Этиология. Нейрофиброматоз 1 типа (НФ1) наследуется по аутосомно-доминантному типу почти с полной пенетрантностью, но варьирующей экспрессией. Тем не менее, положительный семейный анамнез отмечается почти у 50% пациентов, остальные 50% случаев, как полагают, представляют собой новые мутации. Участвует ген супрессоров опухолей, Ген расположен в перицентрической области на длинном плече 17-й хромосомы и имеет чрезвычайно высокую скорость спонтанной мутации более чем с 200 вариантами, известными на настоящий момент.
Продукт гена, нейрофибромин, представляет собой большой цитоплазматический белок, существующий в нескольких формах в различных тканях; он, как полагают, взаимодействует с внутриклеточными цитоплазматическими микротрубочками.
г) Диагностика и лечение нейрофиброматоза 1 типа. Из-за отсутствия общепринятого генетического теста диагноз основан на клинических проявлениях. Они не всегда присутствуют с рождения, проявляясь в разные периоды жизни.
2. Глазные проявления:
— Узелки Пиша: пигментированные (желто-коричневые), гамартомы радужки, легко обнаружить с помощью щелевой лампы. Часто отсутствуют в детстве, узелки Лиша появляются с возрастом и имеются примерно у 94% постпубертатных пациентов. Гистопатологически они представлены массой меланоцитов.
— Другие (необычные) глазные проявления: врожденная глаукома, нейрофибромы век и конъюнктивы, утолщение роговицы, конъюнктивы и цилиарного нерва, астроцитомы сетчатки.
3. Опорно-двигательные нарушения:
— Клиновидная дисплазия крыла основной кости: наиболее типичное костное поражение, приводит к пульсирующему экзофтальму.
— Сколиоз: встречается у 10-20% пациентов, часто проявляется в подростковом возрасте.
— Большеберцовой ложный сустав: может быть устойчивым к лечению с необходимостью ампутации в 80% случаев.
— Другие: истончение коркового слоя кости, гиперплазии конечностей, и (редко) рабдомиосаркома.
4. Неврологические проявления:
— Периферические нейрофибромы: множественные кожные, подкожные и периферические поражения, часто распространяются в торакоабдоминальной области или вокруг соска. Гистологически доброкачественные (злокачественные преобразования редки и могут быть представлены в количестве от нескольких до нескольких тысяч). В основном они представляют собой косметическую проблему с возможностью определенной хирургической коррекции по желанию пациента.
— Плексиформные нейрофибромы: часто поражают большое количество периферических нервов или часть симпатической цепочки с потенциальным обезображиванием (гемигипертрофия руки/ноги) или нарушением функции вовлеченных областей. В целом доброкачественные, но злокачественное преобразование происходит (злокачественная оболочки периферических нервов опухоль: MPNST) в 6% случаев. Для бессимптомных поражений предпочтительна тактика выжидания и наблюдения; результаты лечения с 13-цис-ретиноевой кислотой и интерфероном альфа-2а находятся в стадии оценки.
При симптоматическом течении возможно хирургическое уменьшение/удаление опухоли. Тем не менее, риск местного рецидива высок; помимо этого, многие симптоматические поражения порой включают несколько невральных пучков с высоким риском послеоперационного неврологического дефицита. Для MPNST рекомендуется биопсия с последующей возможной ампутацией конечностей, лучевой терапией и химиотерапией (5-летняя выживаемость: 40%).
— Параспинальные нейрофибромы: наиболее распространенные опухоли, поражающие позвоночник у пациентов с нейрофиброматозом 1 типа (НФ1). Параспинальные нейрофибромы, как правило, возникают из спинальных корешков в шейном и поясничном отделах, они могут врасти в позвоночный канал через межпозвонковые отверстия, в результате чего формируются гантелевидные поражения. Хирургическая резекция показана во всех симптоматических случаях и по рентгенологическим критериям (масс-эффект) для шейно-грудной области (риск вторичной миелопатии в связи со сдавлением спинного мозга).
— Глиомы зрительных путей: наиболее распространенное проявление в центральной нервной системе при нейрофиброматозе 1 типа (НФ1), поражает примерно 15% пациентов. Эта опухоль обычно встречается в детском возрасте с наибольшим риском возникновения в течение первых шести лет жизни; женщины страдают вдвое чаще, чем мужчины. Развитие возможно в любой точке зрительного пути, но чаще всего встречаются прехиазмальные поражения. Приблизительно половина всех оптических глиом протекает бессимптомно, и большинство из них, в том числе симптоматические, редко прогрессируют.
Глазные признаки являются наиболее частыми клиническими проявлениями; у 39% детей с поражениями хиазмы выявляется гипоталамо-гипофизарная дисфункция.
д) Рекомендации:
— Бессимптомные случаи: регулярный офтальмологический осмотр, ежегодно в возрасте до шести лет. При выявлении аномалий проводится МРТ мозга и глазниц.
— Симптоматические случаи: обязательно постоянное наблюдение с офтальмологическими осмотрами и МРТ с контрастным усилением каждые три месяца в течение первых 18 месяцев, с последующими контрольными наблюдениями через определенные интервалы при стабильном состоянии.
Хирургическое лечение ограничено определенными показаниями: у детей с внутриглазничным и прехиазмальным поражениями и закрывающимися глазами (или проптоз глаза с тяжелыми формами нарушения зрения) операция может быть выполнена в косметических целях и для предотвращения распространение опухоли на хиазму. При хиазмальных опухолях вмешательство может потребоваться для уменьшения объема больших опухолей, особенно с кистозным компонентом.
У детей старше трех лет лучевая терапия является вариантом лечения, почти 80% случаев дают стабилизацию/уменьшение опухоли. Тем не менее, и в таких случаях при лучевой терапии существует риск нейрокогнигивных и нейроэндокринных последствий, помимо вызываемых облучением злокачественных новообразований. За последние 15 лет ряд клинических исследований показал, что прогрессирование опухоли может быть приостановлено при использовании одно- и мультиагентной химиотерапии, и, следовательно, химиотерапия в настоящее время предлагается в качест ве терапии первой линии, особенно у более молодых пациентов (моложе трех лет), для которых лучевая терапия противопоказана.
— Аномалии ликворных пространств: некоторое расширение желудочков относительно часто выявляется при нейрофиброматозе 1 типа (НФ1), но его роль в патогенезе задержки психомоторного развития спорна. Действительно, в большинстве случаев расширение желудочковой системы не связано с повышением внутричерепного давления и не является прогрессирующим. Это относится и к расширению субарахноидальных влагалищ корешков спинного мозга. Лишь в исключительных случаях эти менингеальные растяжения могут стать симптоматическими при сжатии или деформации корешков спинного мозга, следовательно, будет необходимо лечение (как правило, шунтирование).
— Другие внутричерепные опухоли: глиомы полушарий встречаются менее чем в 0,5%, а глиомы задней ямки менее, чем в 1% случаев нейрофиброматоза 1 типа (НФ1). Большинство этих опухолей не имеет тенденции к росту. По этой причине, как и для поражений зрительных путей, последовательный мониторинг (клинический и рентгенологический) целесообразен в бессимптомных случаях, а оперативное вмешательство показано для пациентов с симптомами и/или опухолями с тенденцией к росту. Неопознанные яркие включения при МРТ — частое явление у больных с НФ1.
Они представлены областями спонтанной гиперинтенсивности на Т2-взвешенных изображениях, чаще всего с участием базальных ганглиев, в мозжечке, в стволе головного мозга и подкорковом белом веществе. Предполагается, что они представляют собой участки миелинопатии, которые, как правило, разрешаются спонтанно.
— Неврологическая задержка развития: 30-65% пациентов с нейрофиброматозом 1 типа (НФ1) проявляют некоторую степень неспособности к обучению, речевые навыки сохраняются лучше, чем визуальнопространственные.
е) Прогноз. При самом лучшем медицинском обслуживании продолжительности жизни при нейрофиброматозе 1 типа (НФ1) остается примерно на 15 лет меньше, чем у населения в целом. Озлокачествление является одной из основных причин снижения ожидаемой продолжительности жизни у взрослых с НФ1, а общий риск злокачественных осложнений составляет примерно 3-15%.
 Национальный институт здоровья (NIH),
Национальный институт здоровья (NIH),
итоговая конференция по критериям диагностики для нейрофиброматоза (НФ1);
время появления клинических признаков. 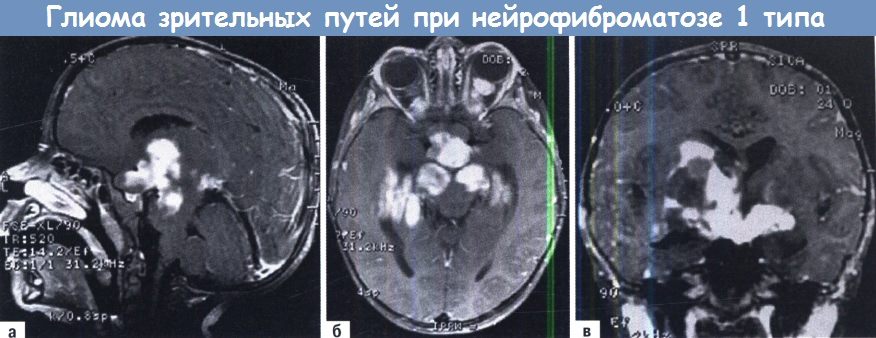 A-В Сагиттальная (А), аксиальная (Б) и коронарная (В) МРТ в режиме Т1 после введения гадолиния 6-месячному ребенку с нейрофиброматозом 1 типа (НФ1) и диффузной глиомой зрительных путей.
A-В Сагиттальная (А), аксиальная (Б) и коронарная (В) МРТ в режиме Т1 после введения гадолиния 6-месячному ребенку с нейрофиброматозом 1 типа (НФ1) и диффузной глиомой зрительных путей.
Как это часто случается у такого рода пациентов, рост опухоли вовлек оба зрительных нерва, хиазму и зрительные тракты.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
НЕЙРОФИБРОМАТОЗ I ТИПА. Проблемы диагностики и лечения
Каковы диагностические критерии нейрофиброматоза? Что является особенностью нейрофиброматоза и затрудняет диагностику? На чем основывается терапия нейрофиброматоза? Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингха
Каковы диагностические критерии нейрофиброматоза?
Что является особенностью нейрофиброматоза и затрудняет диагностику?
На чем основывается терапия нейрофиброматоза?
 Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Заболевание характеризуется выраженным клиническим полиморфизмом, прогрессирующим течением, полиорганностью поражений и высокой частотой осложнений, в том числе приводящих к летальному исходу (развитие сердечно-легочной недостаточности вследствие выраженных скелетных аномалий, злокачественное перерождение нейрофибром и др.).
Механизм развития клинических проявлений неизвестен. Существует предположение, что ген «нф 1» входит в группу генов, подавляющих рост опухолей. Снижение или отсутствие выработки продукта гена — нейрофибромина приводит к диспластической или неопластической пролиферации клеток.
Клиническая диагностика нейрофиброматоза I типа основывается на обнаружении диагностических критериев, рекомендованных Международным комитетом экспертов по нейрофиброматозу. Диагноз может быть поставлен при наличии у больного по крайней мере двух из перечисленных ниже признаков: не менее пяти пятен цвета «кофе с молоком» диаметром более 5 мм у детей препубертатного возраста и не менее шести таких пятен диаметром более 15 мм в постпубертатном периоде; две и более нейрофибромы любого типа или одна плексиформная нейрофиброма; множественные мелкие пигментные пятна типа веснушек, локализованные в крупных кожных складках (подмышечных и/или паховых); глиома зрительного нерва; два и более узелков Лиша на радужной оболочке, обнаруживаемых при исследовании с помощью щелевой лампы; дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных трубчатых костей с наличием псевдоартроза или без него; наличие у родственников первой степени родства нейрофиброматоза I типа по тем же критериям.
 |
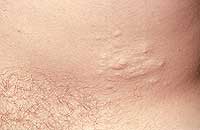
| Рисунок 1. Множественные нейрофибромы |
Особенностью заболевания является специфическая последовательность проявления симптомов в зависимости от возраста пациента, что затрудняет клиническую диагностику нейрофиброматоза I типа в раннем детском возрасте. Таким образом, с рождения или первых лет жизни могут существовать лишь некоторые признаки нейрофиброматоза I типа, такие как крупные пигментные пятна, плексиформные нейрофибромы, скелетные дисплазии. Другие симптомы могут проявиться значительно позднее (к 5–15 годам). При этом степень выраженности клинических проявлений, течение и быстрота прогрессирования нейрофиброматоза I типа у разных больных неодинаковы и колеблются в широких пределах. В настоящее время не установлено, чем обусловлены такие различия.
Нейрофибромы (дермальные, гиподермальные, плексиформные) представляют собой наиболее выраженное проявление болезни Реклингхаузена, их количество иногда достигает нескольких тысяч; плексиформные нейрофибромы могут быть гигантскими, массой более 10 кг. Эти косметические дефекты, как правило, больше всего беспокоят пациентов, даже имеющих системные заболевания. Кроме того, нейрофибромы, особенно плексиформные, связаны с повышенным риском озлокачествления (в 20% случаев, по нашим данным). При локализации в средостении, в брюшной полости, в глазнице они приводят к нарушению функций прилегающих органов. Например, в сентябре 2000 года в отделении наследственных заболеваний кожи ЦНИКВИ был консультирован больной мальчик 8 лет, прибывший из Брянской области в Москву для оперативного лечения по жизненным показаниям; гигантская плексиформная нейрофиброма располагалась в верхнем средостении, деформировала нижнюю 1/3 шеи и являлась причиной затрудненного дыхания и пароксизмальной тахиаритмии.
О развитии нейрофибром известно немногое. Время от времени растет их количество и размеры в ответ на различные стимулы, среди которых ведущее место занимают гормональная перестройка организма: пубертатный возраст, период беременности или после родов, а также перенесенные травмы или тяжелые соматические заболевания. С расширением спектра предлагаемых коммерческих медицинских и косметических услуг значительно увеличилось число обращений больных, указывающих на появление новых опухолей (нейрофибром, неврином, шванном) после ятрогенных вмешательств. Речь идет об удалении опухолей с диагностической или лечебной целью различными методами, в том числе с помощью хирургического иссечения. К ятрогенным осложнениям также приводит назначение физиотерапевтических процедур при лечении различных соматических заболеваний, коррекции скелетных нарушений (всевозможных видов сколиоза, переломов) и нервно-мышечных расстройств (очень часто массаж по различным поводам назначается детям грудного возраста, когда диагностика нейрофиброматоза I типа зачастую невозможна из-за недостаточности клинических проявлений). Но часто заболевание прогрессирует и на фоне кажущегося благополучия. Положение осложняется тем, что у врачей нет возможностей приостановить развитие болезни.
Основной задачей научных исследований представляется разработка методов патогенетического лечения нейрофиброматоза I типа, позволяющих сдерживать появление новых и рост уже имеющихся опухолей, а также предотвращать развитие осложнений.
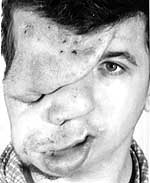 |
| Рисунок 2. Плексиформная нейрофиброма |
В настоящее время для лечения этого заболевания как за рубежом, так и в России используются методы симптоматической терапии, как, например, хирургическое удаление опухолей, коррекция кифосколиоза или лучевая терапия нейрофибром внутренних органов. Кроме того, ученые на Западе сконцентрировались на возможности этиологического лечения, то есть генной инженерии. Особенно далеко это направление продвинулось со времени открытия мутантного гена и расшифровки его первичного продукта — нейрофибромина в 1990 году; на научные изыскания, связанные с этой проблемой, ежегодно выделяются огромные средства.
Первая попытка патогенетического подхода к лечению была сделана V. Riccardi в 1987 г., когда он предложил длительное использование кетотифена (в дозе 2-4 мг в течение 1,5-3 лет) для стабилизации мембран тучных клеток, полагая, что именно дегрануляция этих клеток стимулирует рост опухолей. Однако лечение одним кетотифеном не принесло желаемых результатов: уменьшались субъективные ощущения болезненности и зуда в области нейрофибром, но какого-либо влияния на рост опухолей отмечено не было. Кроме того, при продолжительном приеме препарата наблюдалось снижение количества иммунокомпетентных клеток в периферической крови и ухудшение показателей иммунитета. Существуют различные точки зрения на роль тканевых базофилов в развитии нейрофибром. По мнению некоторых авторов, тучные клетки представляют собой эффекторные клетки противоопухолевого иммунитета. С помощью обычной и электронной микроскопии было показано, что большое количество тканевых базофилов с их активной внеклеточной дегрануляцией наблюдается только на ранней стадии развития нейрофибром. На поздней же стадии, при длительности существования нейрофибром не менее пяти лет, в клеточном матриксе опухолей тучных клеток значительно меньше, дегрануляция их преимущественно внутриклеточная и не сопровождается разрушением клеток.
На основании данных многочисленных исследований нами впервые разработана комплексная методика патогенетической терапии с использованием препаратов из разных групп лекарственных средств. Учитывая то, что клеточный состав нейрофибром в основном представлен шванновскими клетками, фибробластами, тучными клетками и лимфоцитами, а межклеточное вещество в активно растущих, особенно плексиформных, — кислыми мукополисахаридами, для лечения нейрофиброматоза I типа мы выбрали следующие препараты. Стабилизатор мембран тучных клеток, кетотифен, мы назначали по 2-4 мг короткими курсами по два месяца. Чтобы избежать осложнений, в первые две недели приема препарата использовался фенкарол по 10-25 мг три раза в день. В качестве антипролиферативного препарата применялись тигазон в дозе не менее 1 мг на килограмм массы тела или аевит до 600 000 МЕ с учетом переносимости. Также курсами применялась лидаза (мукополисахаридаза) в дозе 32-64 Ед в зависимости от возраста внутримышечно, через день, на курс 30 инъекций.
Вышеуказанные препараты использовались комплексно в различных сочетаниях или в виде монотерапии в зависимости от формы нейрофиброматоза I типа, жалоб, течения, а также возраста и пола больных. Обязательно лечение проводилось в периоды прогрессирования заболевания, то есть при появлении новых опухолей и/или росте уже имеющихся, как правило сопровождающемся зудом или ощущением болезненности в их проекции, а также с целью предотвращения активизации заболевания во время планируемых операций на опухолях. Повторные курсы лечения с интервалами в два месяца назначались при наличии у больных крупных плексиформных нейрофибром или болезненных неврином. При этом, как правило, курсовое применение тигазона (или аевита) в виде монотерапии чередовалось с сочетанным применением стабилизаторов мембран тучных клеток и инъекций лидазы. Предлагаемое лечение хорошо переносилось больными.
В единичных случаях наблюдались незначительное повышение уровней печеночных показателей в повторных биохимических анализах крови при приеме тигазона (у одной больной) и очаговая аллергическая реакция на введение лидазы (у двух пациентов из 60), которая проявлялась воспалением тканей в месте инъекций. В этих случаях препараты отменялись, назначалось симптоматическое лечение.
В результате проводимой терапии нам, как правило, удавалось приостановить прогрессирование заболевания; наблюдалось уменьшение (сморщивание) нейрофибром и неврином вплоть до полного исчезновения некоторых опухолей (особенно активно уменьшаются плексиформные нейрофибромы — на ранней стадии своего развития — и невриномы).
Полученные результаты удовлетворяют исследователей и позволяют рекомендовать вышеуказанную методику патогенетического лечения нейрофиброматоза I типа для повсеместного применения. Разработанная нами комплексная методика патогенетического лечения впервые дает возможность оказать больным медикаментозную помощь.


