Неонатальный сахарный диабет
Основные типы НСД
Транзиторный НСД чаще возникает в течение первых недель, а иногда и дней жизни ребенка. Характерны задержка внутриутробного развития, высокий уровень гликемии и значительная потребность в инсулинотерапии.
В возрасте 3-18 месяцев развивается ремиссия (т. е. исчезновение симптомов) заболевания, однако в старшем возрасте возможен рецидив СД,поэтому даже в случае отмены инсулинотерапии ребенок продолжает наблюдаться эндокринологом.
При перманентном (постоянном) НСД сахароснижающая терапия необходима на протяжении всей жизни ребенка. Реже встречаются синдромальные формы НСД, т.е. сочетание НСД с поражением других органов или систем
Когда можно заподозрить НСД?
В отличие от детей старшего возраста у которых первыми признаками СД являются жажда и частое обильное мочеиспускание (полиурия), наиболее постоянным признаком заболевания у грудных детей является отсутствие прибавки веса или потеря массы тела при адекватном режиме кормления ребенка. Реже родители отмечают появление «крахмальных» пятен на пеленках, липкой мочи, рецидивирующее течение дерматита в паховой области. Иногда НСД выявляется при диспансерном обследовании, например перед вакцинацией, или при сдаче анализов на фоне сопутствующих заболеваний.
Причины развития НСД?
НСД возникает в результате дефекта (мутаций) одного из генов, которые отвечают за нормальное развитие поджелудочной железы, синтез и работу инсулина. Заболевание может передаваться по наследству или возникает спонтанно (т.е. ребенок с НСД может родиться у родителей, не болеющих СД).При этом наиболее частыми причинами перманентного НСД являются мутации в генах KCNJ11, АВСС8 и в гене инсулина (INS). Нужно отметить, что у 25-30% пациентов с дефектами генов KCNJ11 и АВСС8 помимо НСД встречаются неврологические нарушения (эпилепсия, задержка психомоторного и речевого развития) – DEND или iDEND-синдром. Большинство случаев ТНСД связано с аномалиями хромосомы 6q24, реже встречаются дефекты генов KCNJ11, АВВС8 и INS.
Обязательно ли проведение молекулярно-генетического исследования ребенку с НСД?
Если ребенок заболел СД в первые 6 месяцев жизни, проведение генетического исследования обязательно, т. к. при выявлении мутаций в генах KCNJ11 или АВСС8 в большинстве случаев возможно назначение таблетированных сахароснижающих препаратов.
Нужно ли проводить генетическое исследование ребенку, заболевшему СД в 7-12 месяцев жизни, при отсутствии других критериев моногенного СД?
В возрастной группе 7-12 месяцев преимущественно встречается аутоиммунный СД1 типа, генетически обусловленные формы СД выявляются редко. Поэтому детям с дебютом СД от 7 до 12 месяцев жизни предварительно проводится исследование аутоиммунных маркеров СД1 типа. Если антитела отрицательные, решается вопрос о проведении молекулярно-генетического исследования (особенно при отягощенной наследственности по СД). Если антитела положительные, диагностируется СД1 типа и генетическое исследование не проводится.
Глава 8. Неонатальный сахарный диабет (НСД): молекулярно-генетическая гетерогенность и клинический полиморфизм
Термин «неонатальный сахарный диабет» (НСД) включает группу гетерогенных по этиопатогенезу и клинической картине заболеваний, сопровождающихся стойкой (более 2 нед) гипергликемией у детей первых 6 мес жизни [1, 2]. Частота НСД составляет приблизительно 1:250 000 новорожденных в европейских странах [3, 4, 5] и 1:21 196 новорожденных в популяциях с высоким процентом близкородственных браков [6].
Частота заболевания в Российской Федерации не определена.
В 50% случаев НСД является транзиторным (ТНСД) с развитием ремиссии в течение первых месяцев жизни пациента, в остальных случаях отмечается перманентное течение НСД (ПНСД) [7].
На сегодняшний день известно около 20 генов, ответственных за развитие ПНСД. Более половины случаев ПНСД связаны с функциональными дефектами β-клеток в результате мутаций в генах KCNJ11, ABCC8 [8, 9, 10], а также с преждевременным апоптозом β-клеток вследствие дефекта гена INS [11].
У пациентов, рожденных от близкородственных браков, большинство случаев ПНСД связано с гомозиготными мутациями в генах EIF2AK3 (синдром Уолкотт—Раллисон) и GCK [12].
У 25% пациентов с мутациями в гене KCNJ11 [10, 13], а также у некоторых пациентов с мутациями в гене АВСС8 [14] течение НСД осложняется неврологической симптоматикой в виде эпилепсии, выраженной задержки психомоторного и речевого развития, мышечной гипотонии — DEND-синдром.
Также описаны пациенты с промежуточным фенотипом — intermediate DEND (iDEND), характеризующимся умеренной задержкой моторного и речевого развития и отсутствием очагов эпилептической активности на ЭЭГ [10].
Этиопатогенетическая гетерогенность НСД, отсутствие четкой корреляции генотипфенотип определяют необходимость уточнения генетического варианта заболевания. В то же время сложность достижения компенсации углеводного обмена у детей первых месяцев жизни, высокая стоимость лечения, потенциальный риск развития тяжелых сосудистых осложнений уже к моменту достижения пациентом подросткового возраста требуют поиска принципиально новых подходов к лечению данной патологии.
В отличие от аутоиммунного СД1, единственным методом лечения которого на сегодняшний день является инсулинотерапия, большинство пациентов с мутациями в генах KCNJ11 и АВСС8 могут быть успешно компенсированы на фоне приема пероральных сахароснижающих препаратов (производных сульфонилмочевины (СМ)) [15, 16, 17].
В основе этого феномена лежит уникальная способность производных СМ связываться с SUR1-cубъединицей К-каналов, вызывая увеличение секреции инсулина, что с середины прошлого века используется в терапии СД2. Кроме того, назначение производных СМ пациентам с iDEND/DEND-синдромом в ряде случаев приводит к улучшению неврологического компонента заболевания [18].
В настоящей статье приведены результаты генетического обследования 34 пациентов с НСД, получавших лечение в отделении эндокринологии Морозовской детской городской клинической больницы в период с 2008 по 2018 г., обобщен опыт использования производных сульфонилмочевины (ПС) у пациентов с мутациями в генах KCNJ11 и АВВС8.
Ключевые слова: неонатальный сахарный диабет, АТФ-зависимые К-каналы, KCNJ11, ABCC8, DEND-синдром, iDEND-синдром, глибенкламид.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
В исследование было включено 32 пациента (18 мальчиков, 14 девочек) в возрасте от 1 мес до 25 лет с манифестацией СД от 0 до.
Неонатальный сахарный диабет

Неонатальный сахарный диабет – это группа метаболических патологий, которые проявляются гипергликемией и гипоинсулинемией на фоне дисфункции поджелудочной железы. Клинические симптомы включают в себя гипергликемию, полиурию, метаболический ацидоз, кетонемию и кетонурию, дегидратацию, дефицит массы тела. Другие проявления зависят от основной генетической патологии. Диагностика основывается на физикальном осмотре ребенка, лабораторных анализах крови и мочи. Лечение зависит от формы неонатального сахарного диабета. При транзиторном варианте проводится симптоматическая коррекция гипергликемии, при перманентном диабете показана пожизненная инсулинотерапия.

Общие сведения
Неонатальный сахарный диабет (НСД) – это совокупность гетерогенных патологий в неонатологии и педиатрии, для которых характерна гипергликемия и транзиторная или перманентная недостаточность инсулина на фоне дисфункции β-клеток эндокринной части поджелудочной железы. Впервые сахарный диабет у новорожденного описал Kistel в 1852 году. Распространенность данного состояния составляет 1:300-400 тыс. новорожденных. В 55-60% случаев развивается транзиторная форма. Перманентный НСД встречается реже, и, как правило, входит в состав синдромологических патологий. В среднем мальчики и девочки болеют с одинаковой частотой, однако некоторые синдромы (например, IPEX-синдром) более характерны для мужского пола. Тип наследования определенных форм неонатального сахарного диабета также зависит от конкретной генетической аномалии и может быть как аутосомно-доминантным (дефект GK), так и аутосомно-рецессивным (KCNJ11).
Причины
Этиология неонатального сахарного диабета зависит от его клинической формы. Преходящий НСД возникает в результате неполноценного развития β-клеток островков Лангерганса поджелудочной железы. Функционально незрелые клетки неспособны обеспечить адекватную реакцию на повышение гликемии. При этом базовый уровень инсулина в плазме крови может быть нормальным. В большинстве случаев патология развивается спорадически. Также доказана наследственная склонность, связанная с аномалиями длинного плеча VI хромосомы. Причиной возникновения транзиторного неонатального сахарного диабета могут быть мутации генов ABCC8 и KCNJ11, однако дефекты этих же генов в ряде случаев провоцируют развитие перманентной формы.
Персистирующий неонатальный сахарный диабет обусловлен аномалиями структуры β-клеток, всей железы или непосредственно инсулина, из-за чего развивается его абсолютная недостаточность. Как правило, это наследственные дефекты различных генов. Наиболее распространенными вариантами являются гетерозиготные активации мутации генов ABCC8 и KCNJ11. Часто встречаются следующие аномалии, обуславливающие развитие НСД: IPF-1 – гипо- или аплазия поджелудочной железы, GK – отсутствие реакции на уровень глюкозы в крови, EIF2FK3 (синдром Уолкотта-Раллисона) – нарушение процесса синтеза инсулина, FOXRЗ (IPEX-синдром) – аутоиммунное поражение тканей железы. Перманентная форма также может быть проявлением митохондриальных патологий. В некоторых случаях спровоцировать развитие неонатального сахарного диабета может энтеровирусная инфекция, которую мать перенесла в первом триместре беременности.
Классификация
Неонатальный сахарный диабет имеет две основных клинических формы:
Симптомы
Клинические проявления транзиторного и перманентного неонатального сахарного диабета при отсутствии других синдромальных нарушений практически идентичны. При преходящем НСД часто наблюдается задержка внутриутробного развития – дети рождаются с массой тела значительно меньше нормы (ниже 3 перцентиля) для своего срока гестации. Общее состояние ребенка при транзиторной форме нарушено незначительно – пациент малоподвижный, вялый, аппетит снижен или сохранен. Коматозные состояния нехарактерны. Даже на фоне полноценного питания ребенок медленно прибавляет к массе тела. Специфическим признаком неонатального сахарного диабета является выраженная полиурия и дегидратация, часто – резкий запах ацетона изо рта.
Для перманентной формы неонатального сахарного диабета характерны все вышеперечисленные симптомы, но большей интенсивности. Несмотря на это, задержка внутриутробного развития выражена не так сильно. Другие возможные симптомы зависят от того, входит ли НСД в структуру того или иного синдрома. При развитии IPEX-синдрома гипергликемия сочетается другими эндокринными и иммунными нарушениями и целиакнегативной энтеропатией. Клинически это проявляется экземой, хронической диареей, аутоиммунным тиреоидитом, гемолитической анемией. Синдром Уолкотта-Раллисона помимо неонатального сахарного диабета включает почечную недостаточность, нарушения интеллекта, гепатомегалию и спондилоэпифизарную дисплазию.
Диагностика
Диагностика неонатального сахарного диабета включает в себя физикальный осмотр новорожденного, лабораторные и инструментальные методы исследования. Сбор анамнестических данных у матери, как правило, позволяет определить выраженность уже имеющихся проявлений – полиурии, медленной прибавки к массе тела. При объективном обследовании выявляется общая адинамия ребенка, сухость кожных покровов и другие проявления дегидратации. В большинстве случаев у детей наблюдается отставание в физическом развитии и дефицит массы тела.
Лабораторные анализы играют ведущую роль в диагностике неонатального сахарного диабета. При исследовании крови определяется стабильная гипергликемия более 10-11 ммоль/л, повышение уровня кетоновых тел свыше 3 ммоль/л, рНметаболический ацидоз. В анализе мочи можно обнаружить глюкозурию и кетонурию. При проведении пробы по Зимницкому выявляется увеличение суточного диуреза (полиурия) и повышение удельного веса мочи. Инструментальная диагностика в виде УЗД, рентгенографии ОБП, ультрасонографии используется для исключения органических нарушений и определения других проявлений синдромальных патологий. При возможности проводится кариотипирование, генодиагностика с идентификацией дефектных генов.
Дифференциальный диагноз неонатального сахарного диабета осуществляется с гипергликемией новорожденных, гликозурией на фоне массивной инфузионной терапии, внутриутробными инфекциями, воспалительными заболеваниями ЦНС – менингитом и энцефалитом, почечным диабетом, инфекциями кишечного тракта, синдромом мальабсорбции, острыми хирургическими абдоминальными патологиями.
Лечение неонатального сахарного диабета
Терапевтическая тактика при перманентной и транзиторной формах неонатального сахарного диабета существенно отличается. Детям с персистирующим НСД показана заместительная инсулинотерапия, которая дополняется высококалорийным питанием. Терапевтическая схема подбирается индивидуально для каждого ребенка на основе чувствительности к инсулину и уровня глюкозы в крови. Как правило, применяют инсулины как короткого, так и длительного действия. В зависимости от присутствующей синдромальной патологии неонатального сахарного диабета проводится соответствующая коррекция. Например, при мутации гена FOXRЗ назначаются цитостатики, выполняется пересадка костного мозга, а при дефекте KCNJ11 вместо инсулинов используются препараты сульфанилмочевины. Заместительная инсулинотерапия показана на протяжении всей жизни.
У больных с транзиторной формой неонатального сахарного диабета инсулинотерапия используется только при высоких уровнях гликемии, эксикозе, выраженном нарушении общего состояния, дефиците массы тела и ее медленном наборе. На протяжении первых 6-12 месяцев потребность в сахароснижающих препаратах уменьшается, а затем пропадает – наступает полная ремиссия. Контроль над уровнем глюкозы крови и коррекция доз препаратов в зависимости от динамики НСД может проводиться каждые 7 дней или 1 раз в месяц у эндокринолога и педиатра или семейного врача.
Прогноз и профилактика
Прогноз при транзиторной форме неонатального сахарного диабета благоприятный. Как правило, в возрасте от 6 месяцев до 1 года наступает полная клиническая ремиссия. У части детей в дальнейшем может наблюдаться нарушение толерантности к глюкозе. Также существует риск развития аутоиммунного диабета в возрасте 20-30 лет. Прогноз в отношении выздоровления при перманентной форме неонатального сахарного диабета неблагоприятный. Вне зависимости от присутствующих патологий ребенок будет вынужден пожизненно принимать инсулин. Прогноз для жизни при этой форме НСД сомнительный. Исход во многом зависит от наличия тех или иных генетических нарушений. При IPEX-синдроме большинство детей умирают в возрасте до 1 года от тяжелых форм сепсиса.
Специфической профилактики неонатального сахарного диабета не разработано. Неспецифические превентивные меры включают в себя медико-генетическое консультирование семейных пар с оценкой вероятности рождения ребенка с данной патологией. При высоком риске возникновения НСД у будущего ребенка возможно проведение амниоцентеза с последующим кариотипированием.
Неаутоиммунный сахарный диабет у детей
Сахарный диабет (СД) является серьезной проблемой современного здравоохранения во всем мире, влияющей на показатели здоровья, трудоспособности и, в конечном итоге, продолжительности жизни больших групп населения.
Сахарный диабет (СД) является серьезной проблемой современного здравоохранения во всем мире, влияющей на показатели здоровья, трудоспособности и, в конечном итоге, продолжительности жизни больших групп населения. Согласно определению, СД — это заболевание обмена веществ разной этиологии, которое характеризуется хронической гипергликемией, возникающей вследствие нарушения секреции, либо действия инсулина, либо обоих факторов одновременно [1, 2, 3]. Очевидно, что при сходстве финальных метаболических нарушений причины неэффективности действия гормона инсулина могут быть весьма многообразны. Поэтому для выбора адекватной терапевтической тактики важно не только диагностировать собственно диабет, но и максимально детализировать его этиологию.
В детской и подростковой популяции, длительное время считавшейся подверженной заболеваемости исключительно СД 1-го типа (СД 1), проблема дифференциальной диагностики представляется особенно актуальной. Статистика последних лет свидетельствует о возрастании вклада других типов СД в общую структуру заболеваемости СД у детей: СД 2-го типа (СД 2) — до 10%, моногенного СД (МГСД) — 1–3% [2, 3, 4, 5]. Эти данные нельзя считать окончательными в связи с ограничением доступности молекулярно-генетических методов обследования, а также сложившимися стереотипами в отношении диагностики СД у детей.
Между тем диагностические ошибки в установлении этиологии СД способны привести к неверной тактике в выборе терапии основного заболевания, планировании скрининга осложнений и коморбидных состояний и определении отдаленного прогноза. Известные в настоящее время варианты СД у детей, не относящиеся к 1-му типу, в большинстве своем не имеют четких патогномоничных проявлений, что затрудняет их диагностику, определяя необходимость анализа данных в совокупности, включая клинико-лабораторные симптомы, особенности дебюта и течения болезни, ответа на проводимую терапию [5, 6]. В связи с этим мы сочли целесообразным систематизировать современные сведения о неаутоиммунном СД у детей и предложить дифференциальный алгоритм, способный помочь практикующему врачу в диагностическом поиске.
Среди неаутоиммунных вариантов СД у детей можно выделить две основные группы — СД 2 и МГСД.
Сахарный диабет 2-го типа
СД 2 встречается примерно у 10% детей и подростков, больных диабетом. При диагностике следует учитывать особенности этого типа СД, которые должны рассматриваться в комплексе.
СД 2 у молодых имеет этнические особенности, превалируя у небелокожих европейцев, американцев и азиатов, но при этом может встречаться у лиц любой расы. По данным SEARCH for Diabetes in Youth (популяционное исследование среди 10–19-летних жителей США), СД 2 был диагностирован у 33% обследованных афроамериканцев, 22% латиноамериканцев, 40% жителей островов Тихого океана, 76% коренного населения США; при этом среди белых американцев встречался лишь у 6% обследованных. Статистика стран Восточной Азии свидетельствует о высокой заболеваемости СД 2 среди молодых: в Гонконге — до 90%, в Тайване — 50%, в Японии — 60%. Среди молодых жителей США и Европы СД 2 сильно коррелирует с избытком массы тела или ожирением, что не столь характерно для азиатов (в Японии, Индии, Тайване до 30% больных СД 2 детей и подростков имеют нормальный индекс массы тела) [4, 7, 8].
Манифест СД 2 у детей чаще приходится на второе десятилетие жизни (средний возраст — 13,5 лет), совпадая с пиком физиологической пубертатной инсулинорезистентности, которая, в свою очередь, может стать триггером манифестации ранее скрытого СД 2. Варианты клинического манифеста СД 2 широко варьируют. Возможно полное отсутствие клинических симптомов, и тогда СД 2 диагностируется при диспансерном обследовании пациентов из групп высокого риска или случайно. Однако примерно у трети больных в дебюте СД 2 имеет место кетоацидоз, что является частой причиной ошибочного установления диагноза СД 1. Описывается редкий вариант манифеста с развитием тяжелой дегидратации и гиперосмолярной комы с высоким риском летального исхода [9]. Гендерные различия ассоциированы с этническим фактором. Так, в популяции коренных американцев соотношение заболеваемости мальчиков и девочек соответствует 1:4–1:6, а в азиатской популяции подобные различия отсутствуют (1:1). Случаи заболеваемости СД 2 среди родственников, в том числе не первой степени родства, характерны для данного типа диабета. Однако у 15% детей с СД 2, в отличие от взрослых, может отсутствовать отягощенная наследственность [4, 8]. Данный факт наряду с возрастанием роли семейного анамнеза в диагностике СД 1 также может стать причиной ошибок в установлении типа СД у детей.
При СД 2, в отличие от СД 1, отсутствует ассоциация с HLA-маркерами и с аутоантителами.
СД 2, в основе патогенеза которого лежит инсулинорезистентность, может протекать изолированно, но чаще ассоциирован с другими составляющими метаболического синдрома: артериальной гипертензией, дислипидемией, центральным ожирением, acantosis nigricans, овариальной гиперандрогенией, неалкогольным жировым гепатозом (NAFLD), нефропатией. У девочек преждевременное (до 8 лет) изолированное адренархе повышает риск формирования впоследствии овариальной гиперандрогении. Артериальная гипертензия, по разным данным, встречается у 35–75% детей и подростков с СД 2. Нефропатия с симптомами микро- или макроальбуминурии может быть диагностирована как в дебюте, так и при длительном течении СД 2. Есть данные об исходе нефропатии при СД 2 в фокальный сегментарный гломерулосклероз. Дислипидемия характеризуется нарушением соотношения и ростом вклада атерогенных липидных фракций. При СД 2 у детей и подростков, ассоциированным с ожирением, могут иметь место признаки системного воспаления — повышение С-реактивного белка, уровня провоспалительных цитокинов, лейкоцитов, что в целом повышает суммарный кардиоваскулярный риск у взрослых [10, 11, 12, 13].
Установлено, что совокупность кардиоваскулярных факторов риска при наличии инсулинорезистентности и СД определяет высокий риск острых коронарных событий и повышает смертность среди взрослых лиц молодого возраста. Таким образом, СД 2 является серьезной патологией, имеющей особенности в популяции детей и подростков и требующей ранней диагностики, коррекции и своевременного скрининга специфических осложнений и коморбидных состояний [4, 14].
Подходы к ведению больных СД 2 во многом отличаются от таковых при более распространенном у детей СД 1. При сохранении основных компонентов терапии СД (диета, физические упражнения, медикаментозная терапия, обучение), в лечении СД 2 используется «ступенчатый» принцип. Выделяют три терапевтические ступени: диета и физические упражнения; диета, физические упражнения, метформин; диета, физические упражнения, метформин, инсулин.
Стартовая терапия СД 2 определяется клинической картиной и уровнем гликированного гемоглобина (HbA1c). При дебюте с отсутствием клинических симптомов и уровнем HbA1c 5 ммоль/л) приростом гликемии в ответ на углеводную нагрузку в стандартном оральном глюкозотолерантном тесте, сниженным почечным порогом, в связи с чем может определяться глюкозурия при нормогликемии, продолжительным (> 3 лет) периодом «медового месяца». Данные особенности затрудняют раннюю диагностику СД, что влияет на развитие сосудистых осложнений. Тактически пациенты с данным типом СД в дебюте могут получать только немедикаментозную терапию, но в дальнейшем нуждаются в присоединении фармакотерапии. Имеет место высокая чувствительность к препаратам сульфонилмочевины, которые в начале лечения назначаются в дозе, составляющей 1/4 от среднетерапевтической. У детей, нуждающихся в медикаментозной терапии, применяется инсулин [6, 25].
СД вследствие мутации гена HNF-4 альфа (MODY 1) по клиническим характеристикам подобен предыдущему варианту, но без нарушения почечного порога. Может быть диагностирован, когда имеют место клинические проявления MODY 3, но при генетическом исследовании не подтверждается мутация гена HNF-1 альфа [5, 6].
СД вследствие мутации гена глюкокиназы (MODY 2) сложен для диагностики из-за скудности проявлений. Наследуется по аутосомно-доминантному типу. Клинические симптомы, как правило, отсутствуют. Характерными являются длительная «мягкая» гликемия натощак (5–8,5 ммоль/л), высоконормальный или пограничный уровень HbA1c, невысокий прирост гликемии (
И. Л. Никитина, доктор медицинских наук
ФГУ «Федеральный центр сердца, крови и эндокринологии им. В. А. Алмазова» Росмедтехнологий, Санкт-Петербург
Неонатальный сахарный диабет что это
ФГБУ «Эндокринологический научный центр» Минздрава РФ, Москва
ФГУ Эндокринологический научный центр Минздравсоцразвития Российской Федерации, Москва
ФГУ Эндокринологический научный центр Минздравсоцразвития Российской Федерации, Москва
ФГБУ «Эндокринологический научный центр» Минздрава РФ, Москва
Эндокринологический научный центр, Москва
Неонатальный сахарный диабет: эффективность терапии препаратами сульфанилмочевины в зависимости от типа мутаций в гене KCNJ11
Журнал: Проблемы эндокринологии. 2014;60(1): 57-63
Емельянов А. О., Кураева Т. Л., Прокофьев С. А., Медведева Е. Д., Петеркова В. А. Неонатальный сахарный диабет: эффективность терапии препаратами сульфанилмочевины в зависимости от типа мутаций в гене KCNJ11. Проблемы эндокринологии. 2014;60(1):57-63.
Emel’ianov A O, Kuraeva T L, Prokof’ev S A, Medvedeva E D, Peterkova V A. Neonatal diabetes mellitus: the effectiveness of therapy with sulfonylurea preparations depending on the type of mutation in the KCNJ11 gene. Problemy Endokrinologii. 2014;60(1):57-63.
https://doi.org/10.14341/probl201460157-63
ФГБУ «Эндокринологический научный центр» Минздрава РФ, Москва

ФГБУ «Эндокринологический научный центр» Минздрава РФ, Москва
ФГУ Эндокринологический научный центр Минздравсоцразвития Российской Федерации, Москва
ФГУ Эндокринологический научный центр Минздравсоцразвития Российской Федерации, Москва
ФГБУ «Эндокринологический научный центр» Минздрава РФ, Москва
Эндокринологический научный центр, Москва
С начала 20-х годов прошлого столетия инсулинотерапия стала основным методом лечения сахарного диабета (СД) у детей и подростков. Со временем появились данные о неиммунных моногенных формах СД, одной из которых является неонатальный сахарный диабет (НСД), проявляющийся, как правило, в первые 6 мес жизни. НСД встречается с частотой от 1/300 000 до 1/500 000 новорожденных 1. С клинической точки зрения, различают транзиторный (ТНСД) и перманентный (ПНСД) НСД [4].
Молекулярно-генетическая природа НСД оказалась удивительно гетерогенной: в его развитии принимают участие более 10 различных генов [5]. Наиболее клинически значимыми являются мутации в генах KCNJ11 и ABCC8, отвечающих за активность АТФ-зависимых калиевых каналов [6, 7]. АТФ-зависимые калиевые каналы играют центральную роль в глюкозостимулированной секреции инсулина β-клетками поджелудочной железы: секреция инсулина инициируется закрытием каналов и ингибируется их открытием. Калиевые каналы состоят из 4 субъединиц SUR1 и 4 субъединиц Kir6.2 сульфонилмочевинных рецепторов. При поступлении глюкозы в β-клетку с помощью глюкозного транспортера GLUT-2, она метаболизируется, что приводит к накоплению АТФ и закрытию АТФ-зависимых калиевых каналов. Закрытие калиевых каналов ведет к деполяризации клеточной мембраны и открытию кальциевых каналов, т.е. к увеличению концентрации ионов Са++ внутри клетки. Последнее является пусковым механизмом выхода инсулина из клетки. При активирующей мутации в генах калиевых каналов, они остаются открытыми, несмотря на поступление глюкозы в клетку, вследствие чего гипергликемия не приводит к достаточной стимуляции выхода инсулина в кровоток [8].
До недавнего времени единственным методом лечения НСД являлась инсулинотерапия, представляющая большие трудности в связи с малым возрастом пациентов. Потребность в инсулине у каждого ребенка индивидуальна и колеблется от 0,1 до 2,0 ЕД/кг/сут и более. Особые затруднения возникают при необходимости вводить минимальные разовые дозы инсулина (менее 0,5 Ед). Более эффективно непрерывное введение инсулина с помощью инсулиновых помп [12]. Если диагноз СД установлен в возрасте до 6 мес и молекулярно-генетическими методами подтверждены мутации в генах KCNJ11 или ABCC8, возможна альтернативная терапевтическая стратегия. Так как НСД развивается в результате активации АТФ-зависимых калиевых каналов, а пероральные препараты сульфонилмочевины (ППС) приводят к закрытию этих каналов, стимулируя выход инсулина из β-клеток, терапия ППС может быть эффективной альтернативой инъекциям инсулина. Показано, что ППС обеспечивают метаболический контроль и могут заменять инсулин в большинстве случаев ТНСД и ПНСД, вызванных мутациями в генах KCNJ11 и ABCC8. При исследовании больших когорт пациентов с мутациями в этих генах терапия ППС обеспечивала лучший метаболический контроль (снижение уровней гликемии и гликированного гемоглобина) 16. Возникающие транзиторные желудочно-кишечные нарушения и эпизоды гипогликемии устранялись подбором адекватного препарата, дозы и увеличением частоты его введения (например, 2 или 3 раза в день) [18]. Нейромышечные симптомы менее выраженных DEND-синдромов отвечают на терапию глибенкламидом (3-летний мальчик с мутацией I167L [14], взрослый с мутацией G53D [19]). Эти исследования интересны также тем, что доказывают способность ППС проходить через гематоэнцефалический барьер и достаточно эффективно влиять на ЦНС [20].
Международная группа по НСД [21] провела клиническое испытание перорального препарата сульфонилмочевины (глибенкламида) у 49 больных с мутациями в гене KCNJ11. В 90% случаев удалось перейти от инсулина к испытуемому препарату. Продемонстрирован не только более удобный путь введения лекарственного средства, но и значительное улучшение контроля метаболических нарушений. Система длительного мониторирования гликемии показала незначительные колебания постпрандиальной гликемии, а родители отмечали значительное улучшение самочувствия больных детей.
Некоторые тяжелые DEND-мутации нечувствительны к глибенкламиду, даже в очень высоких дозах [22, 23], что побуждает идентифицировать генетический дефект для обеспечения оптимального лечения.
K. Donaghue и соавт. наблюдали 7-летнюю девочку с мутацией в гене KCNJ11, у которой кетоацидоз был отмечен в 7-месячном возрасте, и она оставалась инсулинозависимой, несмотря на максимальные дозы глибенкламида [24]. Эффективные дозы различаются в каждом конкретном случае, и существуют мутации, при которых нет ответа на введение ПСС (табл. 1). 
В последнее время появились указания на связь дозы ППС с типом мутаций (см. табл. 1), что существенно облегчает подбор стартовой дозы терапии [5].
Учитывая, что в нашей стране опыт перевода больных с НСД на препараты сульфонилмочевины невелик, а рекомендации такого перевода неодназначны, приводим собственные наблюдения.
Клинический случай
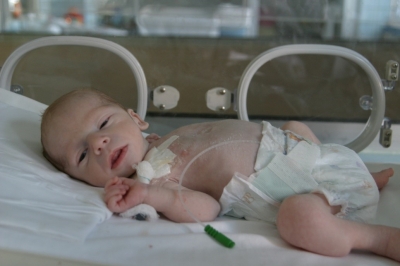
Девочка М., 1 год 6 мес, от 2-й беременности, протекавшей на фоне угрозы прерывания (рис. 1).  Рисунок 1. Девочка М., 1 год 6 мес — ПНСД. За 2 нед до родов мать перенесла ОРВИ. Роды на сроке 39 нед, оперативные (поперечное положение плода). Масса тела при рождении 2960 г, рост 51 см, оценка по шкале Апгар 7-8 баллов, выписана на 6-е сутки. Наследственность: бабушка по материнской линии (61 год) страдает СД2 в течение 5 лет, получает пероральные сахароснижающие препараты. В 2-недельном возрасте у девочки при госпитализации по поводу ОРВИ с подозрением на пневмонию были выявлены гипергликемия и глюкозурия без кетоза. Начата инсулинотерапия в дозе до 1,0 Ед/кг/сут, однако достичь компенсации углеводного обмена не удалось; гликемия колебалась от 2,2 до 30 ммоль/л, отмечались частые гипогликемии.
Рисунок 1. Девочка М., 1 год 6 мес — ПНСД. За 2 нед до родов мать перенесла ОРВИ. Роды на сроке 39 нед, оперативные (поперечное положение плода). Масса тела при рождении 2960 г, рост 51 см, оценка по шкале Апгар 7-8 баллов, выписана на 6-е сутки. Наследственность: бабушка по материнской линии (61 год) страдает СД2 в течение 5 лет, получает пероральные сахароснижающие препараты. В 2-недельном возрасте у девочки при госпитализации по поводу ОРВИ с подозрением на пневмонию были выявлены гипергликемия и глюкозурия без кетоза. Начата инсулинотерапия в дозе до 1,0 Ед/кг/сут, однако достичь компенсации углеводного обмена не удалось; гликемия колебалась от 2,2 до 30 ммоль/л, отмечались частые гипогликемии.
Повторная госпитализация в ЭНЦ в возрасте 1 год 6 мес. Инсулинотерапия при поступлении: новорапид + лантус в соотношении 75:25, суточная доза 0,8-0,9 Ед/кг/сут, суточные колебания гликемии 7,8-22,2 ммоль/л (рис. 2),  Рисунок 2. Девочка М., 1 год 6 мес. Гликемия (по данным CGMS) на инсулинотерапии. уровень HbA 1c 10,0%, секреция С-пептида резко снижена (табл. 2).
Рисунок 2. Девочка М., 1 год 6 мес. Гликемия (по данным CGMS) на инсулинотерапии. уровень HbA 1c 10,0%, секреция С-пептида резко снижена (табл. 2). 
Получены данные о наличии мутации в гене KCNJ11 (R201Н).
Учитывая рекомендуемую для данной мутации дозу (см. табл. 1), произведен перевод на глибенкламид, в стартовой дозе 0,6 мг/кг/сут с последующей титрацией по гликемии под контролем CGMS (доза при выписке 1,2 мг/кг/сут). На этом фоне: снижение дозы инсулина Лантус до 1-0,07 Ед/кг/сут, инсулин НовоРапид отменен; суточные колебания гликемии от 5 до 10 ммоль/л (рис. 3).  Рисунок 3. Девочка М., 1 год 6 мес. Гликемия (по данным CGMS) после назначения глибенкламида. Наблюдалось также восстановление уровня С-пептида при пробе с нагрузкой (табл. 3).
Рисунок 3. Девочка М., 1 год 6 мес. Гликемия (по данным CGMS) после назначения глибенкламида. Наблюдалось также восстановление уровня С-пептида при пробе с нагрузкой (табл. 3). 
Обсуждение
Впервые в нашей стране осуществить успешный перевод пациента с НСД (мутация в гене KCNJ11) на препараты сульфонилмочевины удалось О.А. Малиевскому [25].
В 2010 г. Е.Е. Петряйкина и соавт. описали случай неудачного перевода пациента с НСД (мутация V69М) на глибенкламид (ухудшение гликемического профиля и появление кетоза. В зарубежной литературе нет описания перевода на терапию ППС пациентов с данной мутацией.


