Немалиновая миопатия что это
Немалиновые миопатия (от греч. пета — нить) — нитеобразные включения в мышечных волокнах. При гистологическом исследовании с применением традиционного гематоксилин-эозина трудно прокрашиваются, но легко выявляются при специальных методах окраски. Это не инородные тельца-включения, а структуры, состоящие из избыточного количества материала Z-полос со сходной ультраструктурой.
В их состав входят актин, а-актинин, тропомиозин-3 и белок небулин. Формирование немалиновых структур может служить необычной реакцией мышечных волокон на повреждение, так как они редко обнаруживаются при других заболеваниях. Они в большом количестве присутствуют при врожденной миопатии, известной как немалиновая. Большинство нитей локализуется в пределах миофибрилл (в цитоплазме), при электронной микроскопии в некоторых случаях видны внутриядерные нитевидные структуры, механизм их формирования внутри ядер неизвестен.
Описаны тяжелые младенческие и ювенильные формы немалиновой миопатии. Пациенты с немалиновой миопатией напоминают детей с врожденной диспропорцией типов мышечных волокон, однако заболевание носит более тяжелый характер. Генерализованная гипотония, мышечная слабость, включая слабость мышц бульбарной группы, дыхательные нарушения и резкое снижение мышечной массы — типичные проявления заболевания.
Характерна долихоцефалическая форма головы, высокое готическое нёбо, возможна расщелина нёба. Жевательные мышцы могут быть настолько слабыми, что не удерживают рот в закрытом положении. При рождении может выявляться тяжелая мышечная слабость; некоторые дети умирают в неонатальном периоде. Матери детей с немалиновой миопатией отмечают уменьшение двигательной активности плода во время беременности; новорожденные страдают от гипоксии и дисфагии; в некоторых случаях выявляется артрогрипоз.
Лабораторные исследования немалиновой миопатии. Активность креатинкиназы в плазме крови в пределах нормы. Мышечная биопсия выявляет врожденную диспропорцию типов мышечных волокон или, по крайней мере, преобладание волокон типа I в сочетании с немалиновыми структурами. У некоторых пациентов одинаковые по форме волокна типа I, при этом волокна типа II малочисленны или отсутствуют.
Фокальная дегенерация миофибрилл и повышение активности лизосомных ферментов обнаружены у отдельных пациентов с тяжелой формой заболевания с прогрессирующим течением. Внутриядерные немалиновые структуры коррелируют с наиболее тяжелыми клиническими проявлениями заболевания.
Генетика немалиновой миопатии. Немалиновая миопатия может наследоваться по аутосомно-доминантному и аутосомно-рецессивному типу, описана форма заболевания у девочек с Х-сцепленным доминантными типом наследования. Ген аутосомно-доминантной формы немалиновой миопатии картирован на хромосоме 1 в локусе 1р21-23. Этот ген (ТРМЗ) отвечает за синтез тропомиозина-3 — важного компонента Z-полос. Более распространенная аутосомно-рецессивная форма обусловлена аномалией гена в локусе 2q21.2-q22, определяющего синтез небулина, крупные молекулы которого также служат необходимым компонентом Z-полос.
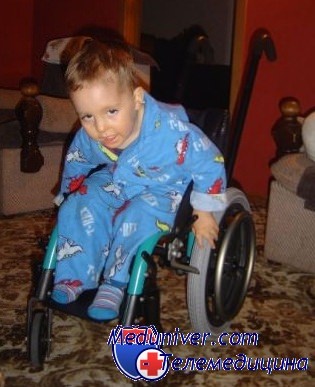
Лечение и прогноз немалиновой миопатии. Терапия носит поддерживающий характер. Выжившие дети прикованы к инвалидному креслу и обычно не способны преодолеть силу тяжести. Отмечается поражение мышц как проксимальной, так и дистальной группы. При хронической дисфагии возможно наложение гастростомы. При ювенильной форме пациенты сохраняют способность к самостоятельному передвижению, повседневная активность пациентов в большей степени сохранена. Слабость обычно не прогрессирует, но у некоторых пациентов двигательные нарушения со временем нарастают или наступает прогрессирующая мышечная слабость.
В редких случаях заболевание осложняется кардиомиопатией, часто наблюдается пневмония.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Немалиновая миопатия

Немалиновая миопатия – группа генетически разнородных наследственных миопатий, общим патогистологическим проявлением которых является образование в мышечной ткани нитевидных структур, что нашло свое отражение в названии патологии (от греческого Nema – нить). Симптомы различных форм состояния могут значительно различаться по своей выраженности – возможны гипотония мышц, мышечная слабость (в том числе дыхательных и мимических мышц), аномалии развития скелета (сколиозы, удлиненная форма черепа, расщелины твердого нёба). Диагностика основывается на оценке статуса больного, изучении биоптата мышечной ткани, молекулярно-генетических исследованиях. Специфического лечения не существует, используется поддерживающая и симптоматическая терапия.
Общие сведения
Немалиновые миопатии (NEM) – совокупность генетических патологий со сходными клиническими проявлениями, которые характеризуются выраженным нарушением структуры мышечной ткани с формированием в ней аномальных нитеподобных или палочковидных структур. Впервые заболевание было описано в 1963-м году двумя независимыми группами исследователей под началом Д. Шая и П. Конена, в последующие годы было выявлено множество генетических разновидностей данного состояния. В зависимости от гена и мутации в нем, немалиновая миопатия может иметь как аутосомно-доминантный, так и аутосомно-рецессивный характер наследования. Встречаемость патологии на сегодняшний день не определена, но, по мнению многих врачей-генетиков, она является одной из наиболее распространенных врожденных миопатий – иногда указываются цифры 1:50 000. При этом достоверно определить этиологию немалиновой миопатии (ответственный за ее развитие ген и характер его дефекта) удается только в половине случаев, что указывает на незавершенность изучения данного заболевания.
Причины и классификация немалиновой миопатии
Причиной развития немалиновой миопатии являются генетические дефекты, приводящие к формированию аномальных форм структурных белков мышечной ткани – в основном, саркомерных протеинов. В результате этого нарушаются процессы сокращения мышц, а также, в ряде случаев, и их формирование, что приводит к появлению врожденных форм данного состояния. На сегодняшний день удалось идентифицировать семь генов, мутации которых ответственны за развитие немалиновой миопатии, однако, как было указано выше, они обуславливают лишь половину всех случаев заболевания. Кроме того, существует форма патологии с отсроченным началом, поражающая в основном взрослых людей – но, как показали более подробные исследования, она обусловлена приобретенными аутоиммунными нарушениями, а не генетическими факторами.
Помимо классификации, созданной на основе данных современной генетики, немалиновая миопатия также разделяется по своим фенотипическим проявлениям на врожденную (типичную, промежуточную и тяжелую разновидности) и ювенильную формы. Взаимосвязь этих двух систем классификации достаточно условна – различные мутации одного и того же гена могут приводить к развитию тяжелых и типичных врожденных форм или ювенильной разновидности заболевания.
NEM-1 – форма немалиновой миопатии, обусловленная мутацией гена TPM3, расположенного на 1-й хромосоме, регистрируется примерно в 2-3% случаев патологии. Ген кодирует одну из цепей медленного альфа-тропомиозина – структурного белка мышечной ткани. К немалиновой миопатии могут приводить различные мутации данного гена с разным типом наследования. Аутосомно-доминантные дефекты становятся причиной развития ювенильной формы заболевания, а аутосомно-рецессивные характеризуются врожденным тяжелым течением патологии.
NEM-2 – наиболее распространенный вариант немалиновой миопатии, составляющий примерно половину от всех генетически идентифицированных случаев данного заболевания. Вызывается дефектом гена NEB, который локализован на 2-й хромосоме и кодирует гигантский белок небулин. Последний функционально связан с тонкими (актиновыми) нитями мышечного волокна. Мутации гена NEB приводят к немалиновой миопатии исключительно врожденного характера с типичным или тяжелым течением.
NEM-3 – вторая по распространенности форма немалиновой миопатии, регистрируемая в 20-25% случаев заболевания с определенной этиологией. Причиной данной формы становятся мутации гена ACTA1, расположенного на 1-й хромосоме. Продуктом его экспрессии является альфа-актин скелетных мышц, один из важнейших структурных компонентов мышечной ткани. Выявлено более 180 типов мутаций данного гена, приводящих к самым разнообразным формам немалиновой миопатии с разным характером наследования, в основном – с врожденными нарушениями. Часть генетических дефектов ACTA1 являются спонтанными мутациями, возникающими de novo, по этой причине иногда возможна зародышевая мозаичность.
NEM-4 – разновидность немалиновой миопатии, составляющая примерно 3-4% всех случаев данного заболевания. Обусловлена дефектом гена TPM2, локализованного на 9-й хромосоме и кодирующего белок бета-тропомиозин, входящий в группу саркомерных протеинов. Мутации этого гена чаще всего передаются по аутосомно-доминантному механизму, имеются указания на возможность спонтанного повреждения гена. Этот генетический дефект проявляется врожденной типичной разновидностью немалиновой миопатии.
NEM-5 – форма немалиновой миопатии, диагностируемая исключительно в закрытых религиозных общинах амиш, где встречаемость этого заболевания достигает 1:500. Вызывается мутацией гена TNNT1, который располагается на 19-й хромосоме и отвечает за белок под названием медленный тропонин Т. Такие генетические нарушения приводят к тяжелой врожденной форме немалиновой миопатии с аутосомно-рецессивным механизмом наследования.
NEM-6 – редкая и малоизученная разновидность немалиновой миопатии, которая предположительно вызывается мутацией гена KBTBD13, локализованного на 15-й хромосоме. Достоверно неизвестно, какой именно белок кодирует данный ген, поэтому его роль в этиологии данного заболевания ставится под сомнение некоторыми исследователями. Предполагают, что его дефекты являются причиной аутосомно-доминантной немалиновой миопатии с легким течением и развитием в детском возрасте (ювенильная разновидность).
NEM-7 – также редкая форма немалиновой миопатии, которая была описана лишь у одной семьи в 2007-м году. Обусловлена дефектом гена CFL2, расположенного на 14-й хромосоме, продуктом его экспрессии является белок кофилин-2 (мышечный кофилин), принимающий активное участие в поддержании стабильного состояния актиновых нитей. Мутации CFL2 приводят к врожденной типичной форме заболевания с аутосомно-рецессивным характером наследования.
Симптомы немалиновой миопатии
По своим клиническим проявлениям все случаи немалиновой миопатии разделяются на две группы – врожденную (которая, в свою очередь, делится на типичную, промежуточную и тяжелую) и ювенильную, для которой характерно появление первых симптомов в детском (8-13 лет) возрасте. Врожденная разновидность встречается намного чаще, может иметь как аутосомно-доминантный, так и рецессивный механизм наследования, тогда как ювенильная практически всегда бывает только аутосомно-доминантной. Больные немалиновой миопатией врожденного типа имеют характерный внешний вид – вытянутое лицо с бедной мимикой, приоткрытый рот, удлиненный череп.
Типичная (умеренная) разновидность является наиболее распространенной, сопровождается миотонией, затруднением при кормлении, антигравитационными движениями. Нарушения дыхания при этой форме немалиновой миопатии выражены незначительно, обычно сводятся к пониженной вентиляции легких по ночам. Слабость скелетных мышц в основном имеет проксимальный характер, прогрессирует крайне медленно, в ряде случаев больные способны передвигаться на костылях или ходить при помощи окружающих.
Промежуточная разновидность врожденной немалиновой миопатии представляет собой более тяжелую форму заболевания. Она характеризуется наличием всех вышеперечисленных нарушений, которые при этом имеют тенденцию к прогрессированию, возможны околосуставные ретракции, искривления позвоночника. Обычно к 6-7 годам больные этой формой немалиновой миопатии полностью утрачивают возможность передвигаться без инвалидного кресла, а к 10-12 годам – самостоятельно дышать, что приводит к летальному исходу. Тяжелая форма немалиновой миопатии проявляется резко выраженной миопатией в неонатальном периоде, затруднением глотания и сосания, в ряде случаев сильно страдает дыхательная мускулатура. Летальный исход наступает в первые месяцы жизни из-за дыхательной недостаточности или респираторной инфекции.
Ювенильная форма немалиновой миопатии ничем себя не проявляет в первые годы жизни ребенка, симптомы заболевания возникают в 8-13 лет. Как правило, обнаруживается мышечная слабость, поражающая как дистальные, так и проксимальные группы мышц, постепенно она прогрессирует, достигая максимальной выраженности к 20-ти годам. В ряде случаев больные ювенильной формой немалиновой миопатии полностью утрачивают возможность ходить, но в отличие от врожденных разновидностей патологии у них отсутствует поражение дыхательной, мимической и жевательной мускулатуры.
Диагностика и лечение немалиновой миопатии
Основными методами диагностики немалиновой миопатии являются изучение наследственного анамнеза пациента, гистологическое исследование мышечной ткани, электромиография и молекулярно-генетические исследования. В анамнезе больного могут определяться случаи заболевания у родственников, отсутствие таких примеров указывает на спорадический характер мутации. Патогистологические изменения в мышечной ткани сводятся к наличию в саркоплазме волокон нитевидных включений – при этом они могут иногда не определяться в мышцах младенцев даже при врожденных формах заболевания и появляться в более старшем возрасте. Иногда при немалиновой миопатии может обнаруживаться также неоднородность толщины мышечных волокон и их частичное разрушение. Электромиография при этом заболевании имеет признаки характерной «миопатической триады» – уменьшение амплитуды и длительности потенциалов действия в сочетании с их полифазностью. При помощи достижений современной генетики можно определить некоторые формы немалиновой миопатии – обусловленные мутациями генов NEB, ACTA1 и TPM2.
Специфического лечения немалиновой миопатии не существует, используют поддерживающую терапию, лечебную физкультуру, гимнастику для сохранения хотя бы минимальной активности мышц и предотвращения суставных контрактур. При нарушении дыхания применяют принудительную вентиляцию легких, питание младенцев в случае нарушений глотания или сосания осуществляют через назогастральный зонд. Если искривления позвоночника или другие скелетные нарушения могут усугубить состояние больного, прибегают к помощи хирургов-ортопедов.
Прогноз и профилактика немалиновой миопатии
В большинстве случаев врожденных разновидностей немалиновой миопатии прогноз заболевания неблагоприятный – у больных имеется высокий риск развития дыхательной недостаточности или респираторной инфекции, что может привести к летальному исходу. Лица с типичной формой патологии могут при адекватном уходе прожить до взрослого возраста, умственное и психическое развитие при этом генетическом заболевании не страдает. Намного более благоприятный прогноз у ювенильной немалиновой миопатии – жизненно важные группы мышц при этом типе патологии не поражаются. Тем не менее, подавляющее большинство больных рано или поздно оказываются прикованными к инвалидному креслу. Лицам с любой формой немалиновой миопатии очень важно следить за своей дыхательной системой и своевременно лечить любые инфекционные процессы в легких.
Врожденная миопатия
Врожденные миопатии — группа редких наследственных заболеваний, при которых поражаются мышцы. Обычно признаки мышечной слабости появляются с рождения. С течением времени они, как правило, медленно прогрессируют. Врожденная миопатия отличается от других наследственных заболеваний, при которых поражаются мышцы, например, от болезни Помпе. 1
Болезнь Помпе — редкая генетическая патология, которая передается по аутосомно-рецессивному типу. Ее признаки могут впервые появиться в любом возрасте — как у новорожденных, так и у пожилых людей. Из-за наследственной мутации в организме развивается недостаточность фермента кислой альфа-глюкозидазы, или кислой мальтазы, которая расщепляет гликоген. 1
Нерасщепленный гликоген накапливается в мышцах, что приводит к их поражению. Классические симптомы болезни Помпе — выраженное снижение мышечного тонуса, постоянно усиливающаяся мышечная слабость, увеличение размеров сердца (при развитии заболевания у детей до 1 года), одышка. Болезнь постоянно прогрессирует, однако при своевременном лечении можно значительно улучшить состояние и прогноз жизни пациента. 1
Так же, как и болезнь Помпе, врожденные структурные миопатии связаны с генными мутациями. Какие виды миопатий встречаются чаще, как они проявляются и лечатся?
Классификация и симптомы
Выделяют несколько видов классических патологий. Самые распространенные формы 2 :
Симптомы патологий могут быть различными. Один из главных признаков – мышечная слабость, которая появляется с рождения. Тем не менее, известны случаи, когда слабость появляется во взрослом возрасте, хотя это происходит нечасто. 2
Слабость мышц может возникать внутриутробно, из-за чего плод недостаточно активен. Наибольшая гипотония мышц обычно отмечается в тазовом поясе и ногах. Мышцы рук и плечевого пояса страдают меньше. 2
При миопатии у детей могут выявляться скелетные аномалии — вывих бедра, полая стопа, кифосколиоз, контрактуры крупных суставов и другие. Иногда могут возникать мышечные боли во время или после физической нагрузки и даже в состоянии покоя. 2
Выраженность симптомов может быть очень незначительной, настолько, что иногда человек даже не подозревает, что болеет. Но некоторые патологии проявляются конкретными признаками. К примеру, при немалиновой миопатии развивается сколиоз, килевидная грудная клетка, а лицо приобретает удлиненную, продолговатую форму. 2
Особенности распространенных форм врожденной миопатии
Болезнь «центрального стержня» связана с генетическими нарушениями молекул, которые перераспределяют кальций в мышцах при сокращении. Это приводит к нарушениям в центральных зонах скелетно-мышечных волокон, внутри большинства из них образуются «стержни»-тяжи. Классические признаки заболевания – мышечная слабость, снижение тонуса мышц. 3
Многостержневые миопатии характеризуются наличием множественных участков мышечного волокна, имеющих сниженную окислительную активность. Болезнь обычно проявляется в младенческом возрасте с мышечной гипотонии и задержки формирования двигательных навыков на несколько месяцев. К примеру, ходить ребенок с многостержневой миопатией начинает на 4-6 месяцев позже, чем здоровый. 4
Центроядерная миопатия — заболевание, при котором в мышечных волокнах образуется множество центральных ядер. В зависимости от типа наследования может проявляться в младенческом, подростковом и взрослом возрасте. В первом случае прогноз наиболее неблагоприятен. 5
Немалиновая миопатия характеризуется наличием в цитоплазме мышечных волокон включений в форме нитей. Заболевание проявляется мышечной слабостью, снижением мышечного тонуса, задержкой в приобретении двигательных навыков у детей. 6
Диагностика
При появлении мышечной слабости важно обратиться к врачу, чтобы выяснить причину и диагностировать патологию. При подозрении на врожденную миопатию врач может назначать комплексное обследование, в которое входят:
Лечение
Существует только симптоматическое лечение заболевания. В первую очередь оно направлено на поддержание дыхания, поскольку эти расстройства считаются одними из самых тяжелых осложнений. Для коррекции дыхательных нарушений назначают лечебную физкультуру, массаж, другие лечебные процедуры. 2
Для коррекции контрактур, нарушений осанки рекомендуют занятия лечебной физкультурой, физиотерапевтическое лечение, иглорефлексотерапию. А вот лекарственных препаратов, способных эффективно облегчить течение этих патологий, не существует. 2 В отличие от болезни Помпе, для лечения которой доступна патогенетическая терапия. 1
Прогноз
Различные виды мышечных миопатий имеют разную степень тяжести: от выраженной, приводящей к инвалидизации, до минимальных проявлений. Прогноз зависит от вида патологии. Важный критерий прогноза — скорость нарастания мышечной слабости: чем медленнее она прогрессирует, тем лучше прогноз.
Немалиновая миопатия что это
Врожденные миопатии клинически характеризуются гипотонией и слабостью, обычно наблюдаемыми с рождения, и морфологическими изменениями при гистологических исследованиях и/или электронной микроскопии. Гистопатологические признаки мышечной дистрофии отсутствуют, уровень креатинкиназы (КК) нормальный или слегка повышен, результаты ЭМГ нормальные или свидетельствуют о миопатии. Отмечается широкая вариабельность степени тяжести клинических проявлений внутри каждой формы миопатии и значительное наложение клинической картины других нейромышечных расстройств, в том числе мышечных дистрофий, метаболических миопатий и спинальных мышечных атрофий, а также синдрома Прадера-Вилли, который тоже может дебютировать у новорожденных выраженной гипотонией («гипотоничный младенец»).
Некоторым врожденным миопатиям дано четкое клиническое, морфологическое и генетическое определение. Некоторые определены по специфическим структурным нарушениям, но до сих пор не подтверждены генетическими исследованиями, что препятствует их выделению в отдельную нозологическую форму.

Несмотря на успехи молекулярной генетики, диагноз обычно ставится на основании результатов исследования биоптата мышечной ткани, и, поскольку, в отличие от мышечных дистрофий, клинические картины различных расстройств накладываются одна на другую, направление пациента сразу на анализ ДНК в настоящее время не имеет практического значения.
В настоящее время нет способов излечения врожденных миопатий, но междисциплинарный подход к ведению таких больных может значительно повысить как качество, так и сроки их жизни — в частности, физио- и трудотерапия, ортопедическое лечение, контроль нарушений дыхания и коррекция трудностей кормления. Иногда болезнь протекает относительно доброкачественно, но часто больной становится тяжелым инвалидом, или даже умирает в детском возрасте. Для нескольких из этих расстройств отсутствуют методы пренатальной диагностики. Вебсайт, содержащий информацию о применяемых генетическом тестах: www.geneclinics.org.
а) Немалиновая миопатия. «Типичная врожденная» форма немалиновой миопатии обычно проявляется при рождении или в первый год жизни гипотонией, слабостью и трудностями при кормлении. У некоторых детей она проявляется позже задержкой двигательного развития, утиной походкой или нарушениями речи, тогда как в некоторых случаях болезнь не проявляется до достижения взрослого возраста. Часто наблюдается слабость мышц лица. Одинаково часто развивается слабость дистальных и проксимальных отделов конечностей. Всегда поражается дыхательная мускулатура, и хотя гиповентиляция может протекать субклинически, нередко возникает необходимость в активной искусственной дыхательной поддержке. Патология сердца наблюдается редко. Заболевание часто протекает статично или прогрессирует очень медленно, и большинство пациентов смогут вести активный образ жизни (Ryan et al., 2001).
Тяжелая врожденная форма немалиновой миопатии проявляется с рождения тяжелой степенью гипотонии и мышечной слабости, малым объемом спонтанных движений, нарушениями сосания и глотания и дыхательной недостаточностью. Эти больные часто умирают в первые недели или месяцы жизни из-за дыхательной недостаточности или рецидивирующей пневмонии, хотя некоторые пациенты выживают и обретают способность ходить (Ryan et al., 2001).
Диагностические признаки немалиновой миопатии — это наличие четких палочковидных включений — немалиновых тел в волокнах скелетной мускулатуры. Палочки не видны при окраске гематоксилином-эозином, но становятся заметными как красные или лиловые структуры на сине-зеленом фоне миофибрилл при трехцветной окраске в модификации Гомори. Считается, что эти включения являются производными боковых расширений Z-линии. Дополнительные патологические признаки включают преобладание волокон I типа, атрофию и/или гипертрофию волокон (Ryan et al., 2003).
Немалиновая миопатия может наследоваться как аутосомно-доминантное, аутосомно-рецессивное или спорадическое состояние (включая новые доминантные мутации). За последние девять лет были открыты мутации пяти новых генов. Все эти гены кодируют протеиновые компоненты тонких мышечных филаментов: α-тропомиозинмедленный, небулин, скелетный α-актин, β-тропомиозин и тропонин Т. Рецессивные мутации небулина, вероятно, вызывают заболевание почти в 50% случаев. Спорадические или новые доминантные мутации в гене скелетного а-актина (AСТА1) вызывают 20-25% случаев (и почти 50% смертей новорожденных).
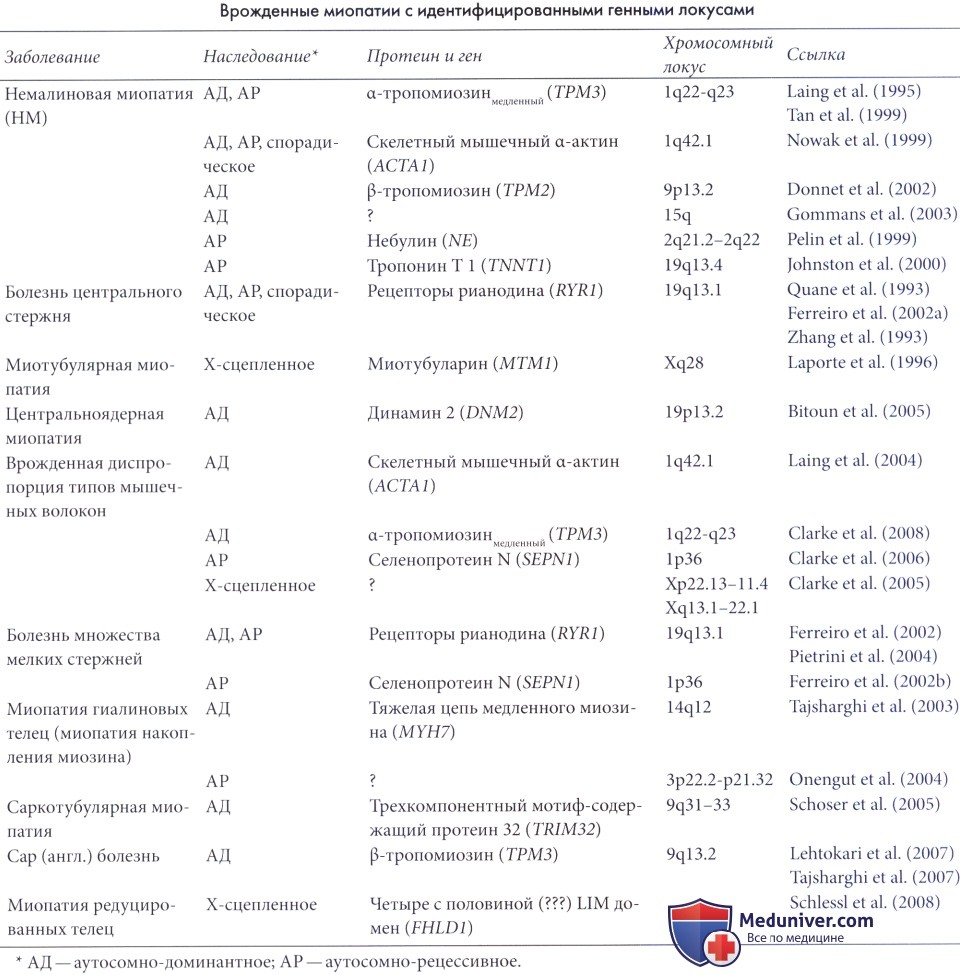
 Немалиновая миопатия. Тяжелая неонатальная форма (слева).
Немалиновая миопатия. Тяжелая неонатальная форма (слева).
Полутонкий срез окрашенный толуидиновым синим. Палочкообразные структуры видны в многочисленных, чаще всего атрофичных, ангулярных волокнах.
Детская форма (справа), электронная микроскопия (х47,000). Эозинофильные неорганизованные структуры протеина Z-полосы.
б) Болезнь центрального стержня (БЦС). Болезнь центрального стержня (БЦС) обычно манифестирует в младенческом возрасте слабостью и гипотонией, проявляясь, прежде всего, поражением мышц проксимальных отделов/нижних конечностей и малой мышечной массой. Большинство пациентов способно к самообслуживанию. Двигательное развитие обычно протекает с задержкой, но мышечная слабость обычно не прогрессирует или прогрессирует медленно. Может наблюдаться небольшая слабость мышц лица и шеи. Экстраокулярные мышцы обычно интактны. Часто развиваются мышечноскелетные деформации, такие, как кифосколиоз, врожденный вывих бедра, полая стопа или плоскостопие и деформации грудной клетки. Не характерна выраженная дыхательная недостаточность, патология сердца встречается редко, состояние интеллекта не нарушено (Shuaib et al., 1987).
Хотя у большинства пациентов развивается «типичный» клинический фенотип, центральные стержни могут встречаться и при отсутствии клинических проявлений у пациентов с невыраженной слабостью и легкими скелетными деформациями; единственным проявлением заболевания может быть также повышение уровня сывороточной КК (Shuaib et al., 1987; Quinlivan et al., 2003). Недавно, после выявления детей с доминантным или рецессивным БЦС, клинический спектр БЦС был расширен; у них болезнь проявлялась артрогрипозом, тяжелой мышечной слабостью и дыхательной недостаточностью в неонатальном периоде (Romero et al., 2003). У нескольких пораженных детей также описаны офтальмоплегия и птоз. Таким пациентам с самого рождения может понадобиться респираторная поддержка, но у выживших детей с возрастом могут значительно улучшиться дыхательные функции и увеличиться мышечная сила (Romero et al., 2003).
Болезнь центрального стержня (БЦС) традиционно считалась аутосомно-доминантным заболеванием с вариабельной пенетрантностью. Относительно часто встречаются мутации de novo, особенно в тяжелых случаях (Ferriero et al., 2002а; Romero et al., 2003). Большинство случаев болезни центрального стержня вызваны мутациями гена RYR1 хромосомы 19ql3.1 (Quane et al., 1993; Zhang et al., 1993; Ferriero et al., 2002a; Romero et al., 2003). Ген RYR1 кодирует рианодиновый рецептор, регулирующий выход кальция из саркоплазматического ретикулума в саркоплазму, вызывая мышечное сокращение. БЦС аллельна злокачественной гипертермии, и поэтому необходимо соблюдать осторожность при анестезиологическом обеспечении всех пациентов с мышечной слабостью неясной этиологии и вероятным аутосомно-доминантным наследованием.
У некоторых пациентов со злокачественной гипертермией при исследовании биоптата мышцы выявляются центральные стержни, хотя мышечная сила у них остается нормальной.
Характерная аномалия при болезни центрального стержня (БЦС) — наличие центральных стержней: единичных, четко очерченных округлых зон в центре большинства волокон I типа. Стержни являются зонами сниженной окислительной активности ферментов, негативными при окраске на фосфорилазу и гликоген (Shy и Magee 1956; Quinlivan et al., 2003). В стержнях отсутствуют митохондрии и саркоплазматический ретикулум (Quinlivan et al., 2003). В большинстве случаев преобладают волокна I типа.
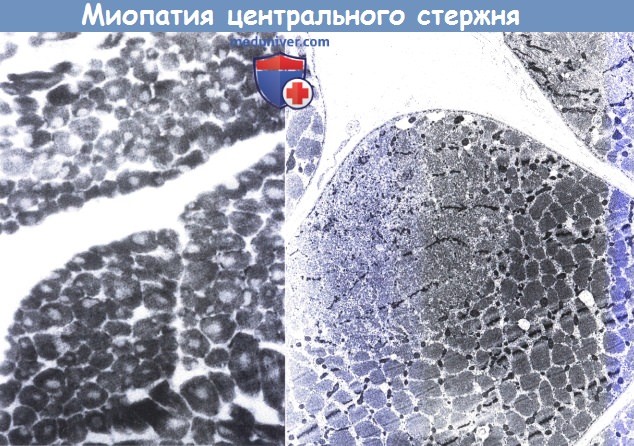 Миопатия центрального стержня.
Миопатия центрального стержня.
Обратите внимание на однотипность волокон (I типа) и наличие четкой центральной зоны, где отсутствует ферментная активность практически во всех волокнах (слева) (НАД-Нх 150).
При электронной микроскопии (справа) видно, что в «зоне стержня»
(которая в донном конкретном волокне смещена к периферии, а не находится в центре) отсутствуют нормальные мышечные фибриллы и митохондрии; также видна дезорганизация миофиламентов (х23,000).
в) Миотубулярная и центронуклеарная миопатия. На основании клинических и генетических признаков выделяют три группы миотубулярных (центронуклеарных миопатий). Лучше других клинически и генетически определена Х-сцепленная рецессивная форма. Заболевание обычно манифестирует во внутриутробном периоде, течение беременности осложняется многоводием. В анамнезе по материнской линии могут быть случаи невынашивания и смерти новорожденных мужского пола. У пораженных младенцев мужского пола с рождения наблюдаются тяжелые общая гипотония и мышечная слабость, лицевая диплегия, трудности при дыхании и кормлении. Дополнительные признаки включают в себя тонкие ребра, контрактуры тазобедренных и коленных суставов, толстые отечные веки, офтальмоплегию и крипторхизм. Многие больные мальчики умирают в неонатальном периоде, но небольшое число больных живет в течение длительного времени; последним часто требуется постоянная дыхательная поддержка (McEntagart et al., 2002).
Ген Х-сцепленной миотубулярной миопатии, МТМ1, локализуется в хромосоме Xq28 (Laporte et al, 1996), считается, что он участвует в пути сигнальной трансдукции, необходимом в процессе позднего миогенеза. Основное заболевание, с которым проводится дифференциальная диагностика,— врожденная миотоническая дистрофия — исключается на основании клинической картины и генетических исследований.
Аутосомная форма миотубулярной миопатии известна как центронуклеарная миопатия и встречается крайне редко. Аутосомно-рецессивная форма центронуклеарной миопатии обычно манифестирует в младенческом возрасте или раннем детстве респираторным дистрессом, гипотонией, тихим криком и нарушением сосания. Часто наблюдаются офтальмоплегия, птоз и лицевая диплегия. Клиническое течение характеризуется задержкой двигательного развития, медленно прогрессирующей слабостью, развитием сколиоза. По достижении подросткового возраста многие пациенты прикованы к инвалидному креслу (Jeannet et al., 2004). Хромосомный локус рецессивной формы центронуклеарной миопатии до сих пор не известен. Аутосомно-доминантные и спорадические формы болезни обычно дебютируют в детстве, подростковом или взрослом возрасте, и имеют значительно более легкий фенотип и более медленно прогрессирующую мышечную слабость и истощение; часто наблюдаются птоз и ограничение подвижности глаз.
Мутации, вызывающие заболевание, были недавно обнаружены в гене, кодирующем динамин 2 (Bitoun et al., 2005).
Характерные гистологические признаки — это преобладание маленьких волокон I типа с центрально расположенными ядрами, напоминающими фетальные миотубы (North, 2004). Обычно наблюдается агрегация митохондрий в центре мышечного волокна, сопровождаемая плотным окрашиванием окислительных ферментов и отсутствием окрашивания миозин аденозин трифосфатазы (АТФ-азы). Радиальное расположение саркоплазматических тяжей при окислительном окрашивании может быть характерным признаком этого заболевания (Jeannet et al., 2004). Облигатное носительство Х-сцепленных форм миотубулярной миопатии обычно протекает бессимптомно, но до 50% таких носителей имеют типичные изменения в биоптате мышечной ткани.
г) Многострежневая миопатия. Многострежневая миопатия — клинически гетерогенное заболевание. Примерно у 75% пациентов наблюдается «классическая» форма расстройства, манифестирующая в течение первого года жизни. Больные младенцы гипотоничны и слабы, отмечается гиперподвижность суставов и задержка двигательного развития. Наблюдается слабость преимущественно аксиальной мускулатуры; в наибольшей степени поражаются мышцы-сгибатели шеи, что приводит к снижению или отсутствию контроля головки у младенцев. Часто развиваются кифосколиоз и ригидность позвоночника, которые могут быстро прогрессировать в периоды быстрого роста скелета, особенно в подростковом возрасте. Этим процессам может сопутствовать дыхательная недостаточность, которая также может развиться в короткие сроки (Ferreira et al., 2000; Jungbluth et al., 2000).
Почти у двух третей пациентов с классической формой болезни множества мелких стержней в позднем подростковом периоде или в раннем взрослом возрасте развивается дыхательная недостаточность, хотя многие из них сохраняют способность к самообслуживанию (Ferreira et al., 2000). Часто наблюдается слабость мышц лица, но экстраокулярные мышцы сохранны. Интеллект нормальный. Болезнь множества мелких стержней также может проявляться легкой слабостью дистальных отделов верхних конечностей с амиотрофией кисти и выраженной гиперподвижностью суставов. Поражение нижних конечностей не тяжелое, отмечается слабость в проксимальных мышцах поясницы и таза. При этой легкой форме выраженность сколиоза и нарушения дыхания минимальны или вовсе отсутствуют (Ferriero et al., 2000).
Редко болезнь множества мелких стержней может проявляться наружной офтальмоплегией и слабостью мышц проксимальных отделов конечностей (Jungbluth et al., 2000) или в неонатальном периоде множественным врожденным артрогрипозом (Ferriero et al., 2000).
Мелкие стержни выявляются в препаратах на АТФ-азу в виде множественных фокальных дефектов окислительной активности ферментов в большинстве волокон; они встречаются как в волокнах 1, так и 2 типа (Ferriero et al., 2000; Jungbluth et al., 2000). Этим изменениям сопутствует увеличение вариабельности размера волокон, наличие центральных ядер, гипотрофия и преобладание волокон I типа (Jungbluth et al., 2000). Дистрофические изменения иногда наблюдаются у пациентов с болезнью множества мелких стержней, вызванной мутациями SEPN1 (Ferriero et al., 2002b).
Было выявлено, что мутации двух генов вызывают примерно 50% случаев болезни множества мелких стержней. Рецессивные мутации SEPN1, гена селенопротеина N, вызывают 30% случаев, особенно часто — случаи с ранним тяжелым кифосколиозом и высоким риском развития дыхательной недостаточности (Ferriero et al., 2004). Различные мутации того же гена вызывают мышечную дистрофию ригидного позвоночника (Moghadazadeh et al, 2001), десмин-связанную миопатию с тельцеподобными включениями Mallory (Ferriero et al. 2004) и врожденную диспропорцию типов волокон (Clarke et al., 2006). Клинические картины всех этих состояний очень похожи, несмотря на различные патологические изменения мышц.
Рецессивные мутации в гене мышечного рианодина (RYR1) имеются у небольшого количества пациентов (Pietrini et al., 2004), и опубликовано единственное сообщение о доминантной мутации RYR1, вызвавшей дебютировавшую у взрослого болезнь множественных мелких стержней (Ferriero et al., 2002а). Пациенты с болезнью множественных мелких стержней, вызванной мутацией RYR1, имеют высокий риск развития злокачественной гипертермии (Guis et al., 2004).
д) Врожденная диспропорция типов мышечных волокон (ВДТМВ). Термин врожденная диспропорция типов мышечных волокон (ВДТМВ) впервые был впервые предложен Brooke в 1973 г. в докладе о группе из 12 детей с гипотонией, признаками врожденной миопатии, и определенными гистологическими аномалиями. Определяющей аномалией при световой микроскопии было различие в размерах мышечных волокон 1 типа (медленное сокращение) и 2 типа (быстрое сокращение), первые оказались минимум на 12% меньше последних (Brooke и Engel, 1969). Гистологические изменения были названы диспропорция размеров волокон (ДРВ).
Диспропорция размеров волокон (ДРВ) как вторичный феномен может наблюдаться и при другой клинической картине при самых разнообразных скелетных и нейромышечных расстройствах, включая расстройства ЦНС, периферическую нейропатию, мышечную дистрофию ЦНС, периферическую нейропатию, мышечную дистрофию и другие врожденные миопатии, такие, как немалиновая миопатия, болезнь множественных мелких стержней и центронуклеарная миопатия. В результате ВДТМВ — это диагноз исключения, который выставляется пациентам с клиническими проявлениями врожденной миопатии, у которых единственной выявленной гистологической аномалией является уменьшение размера мышечных волокон 1 типа по сравнению с волокнами 2 типа (Bodensteiner, 1994; Clarke и North, 2003).
У большинства пациентов с ВДТМВ с рождения наблюдается гипотония, мышечная слабость, уменьшающаяся с возрастом, маленький рост, низкая масса тела, множественные контрактуры суставов (особенно врожденный вывих бедра и эквиноварусная косолапость), сколиоз, длинное тонкое лицо, высокое небо и изогнутая верхняя губа (Brooke, 1973; Innaccone et al., 1987). Реже у пациентов с ВДТМВ с детства отмечается медленно прогрессирующая слабость, офтальмоплегия, поражения глотки и сердца, снижение интеллекта и тяжелые прогрессирующие двигательные нарушения с кифосколиозом и дыхательной недостаточностью (Iannaccone et al., 1987; Haltia et al., 1988; Torres и Moxley, 1992; Akiyama и Nonaka, 1996; Banwell et al., 1999).
Врожденная диспропорция типов мышечных волокон (ВДТМВ) является генетически гетерогенным состоянием, сообщалось о семьях с аутосомно-доминан гным и рецессивным наследованием. У пациентов с тяжелой манифестировавшей с рождения мышечной слабостью и поражением дыхательной мускулатуры были выявлены возникшие de novo доминантные мутации гена, кодирующего скелетный а-актин (АСТА1), также связанные с немалиновой миопатией (Laing et al., 2004). Также были выявлены рецессивные мутации гена, кодирующего селенопротеин N (SEPN), связанные с болезнью множественных мелких стержней (Clarke et al., 2006). В дополнение, в семье, в которой у мужчин развивалась тяжелая летальная миопатия, а у женщин-носителей — слабость мышц лица и птоз, был идентифицирован локус Х-хромосомы (Clarke et al., 2006).
е) Миопатия гиалиновых телец (миопатия накопления миозина). Эта редкая врожденная миопатия характеризуется генерализованной мышечной слабостью или слабостью мышц проксимальных отделов и манифестирует в младенческом возрасте, в детстве или, редко, во взрослом возрасте (Masuzugava et al., 1997; Tajsharghi et al., 2003). Гиалиновые тела представляют собой большие зоны, лишенные саркомеров, локализующиеся на периферии мышечного волокна, содержащие мелкогранулированный материал, где после обработки кислотой выявляется активность миофибриллярной АТФ-азы. Сопутствующие патологические изменения включают в себя диспропорцию типов мышечных волокон, преобладание волокон 1 типа.
Аутосомно-доминантная миопатия гиалиновых тел (накопления миозина), как было недавно выяснено, вызывается мутациями гена, кодирующего тяжелую цепь медленного миозина MYH7 (Tajsharghi et al., 2003), эти мутации также вызывают семейную гипертрофическую кардиомиопатию (Bonne et al., 1998), и дистальную миопатию Лаинга (Meredith et al., 2004). Рецессивная миопатия гиалиновых телец сцеплена с локусом третьей хромосомы (Onengtit et al., 2004).
ж) Саркотубулярная миопатия. Это редкое рецессивное заболевание, характеризующееся вакуолизацией в мышечных волокнах, видной при световой микроскопии (Jerusalem et al., 1973). Вакуоли связаны с мембраной и, как оказывается, развиваются из саркотубулярной системы. Клинически заболевание проявляется непрогрессирующей слабостью проксимальных отделов, вариабельным поражением сгибателей шеи, мышц лица и дыхательной мускулатуры, возникающих с рождения. У сиблингов описаны значительно различающиеся по тяжести и времени проявления вызываемой упражнениями миалгии и слабости мышц проксимальных отделов (Muller-Felber et al., 1999). Мутации, вызывающие заболевание, были идентифицированы в гене TRIM32, который также связан с одной из форм конечностно-поясничной мышечной дистрофии (КПМД 2Н).
з) Синдром Маринеску-Шегрена (СМШ). Синдром Маринеску-Шегрена (СМШ) — давно описанное аутосомно-рецес-сивное мультисистемное заболевание, проявляющееся с младенческого возраста атаксией вследствие мозжечковой дисплазии, умственной отсталостью слабой или средней степени, миопатией, мышечной слабостью и маленьким ростом. Описаны дополнительные симптомы, а именно скелетные аномалии и гипергонадотропный гипогонадизм. Заболевание обычно становится заметным на поздних стадиях из-за поражения мышц, характеризующегося генерализованными гипотонией и слабостью, и миопатической ЭМГ (Zimmer et al., 1992). Электронное микроскопическое исследование биоптата мышечной ткани выявило плотную мембранозную структуру, окружающую ядра мышечных волокон и являющуюся характерным ультраструктурным признаком СМШ (Sewry et al., 1988).
После идентификации вызывающей заболевание мутации гена SIL1, ключевого регулятора основных функций эндоплазматического ретикулума, СМШ был включен в число болезней дисфункций эндоплазматического ретикулума, что указывает на роль этой органеллы в развитии мультисистемных заболеваний (Senderek et al., 2005).
Редактор: Искандер Милевски. Дата публикации: 14.1.2019


