Спинальная мышечная атрофия: что это такое и как ее вылечить
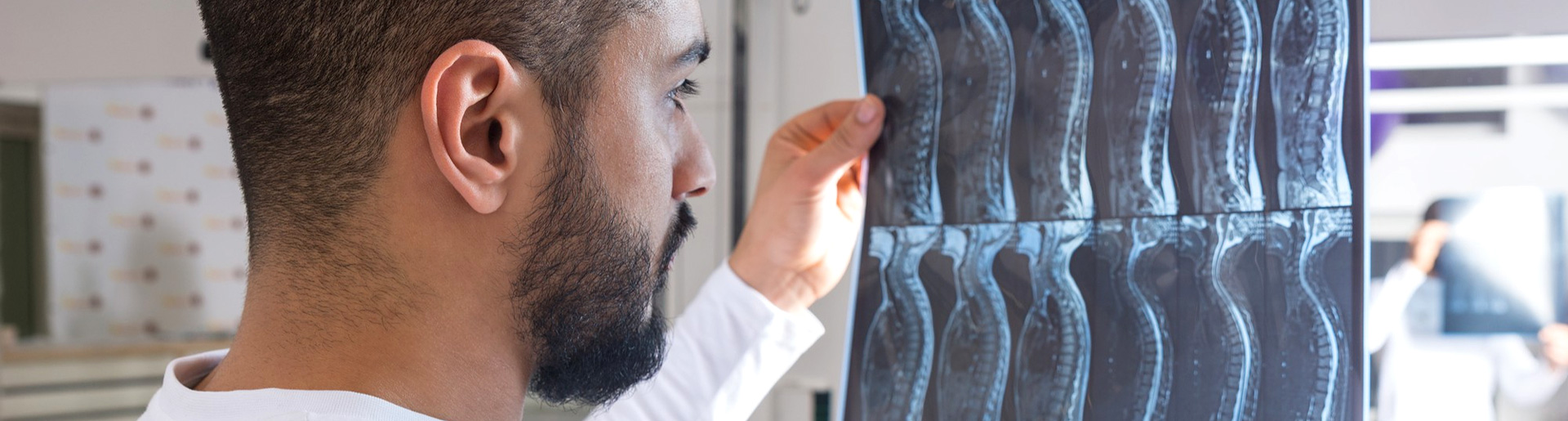
О спинальной мышечной атрофии у детей говорят все чаще, обычно — в контексте появления долгожданных лекарств против нее. Можно ли спасти всех больных и что для этого требуется?
Что такое СМА?
Спинальная мышечная атрофия (СМА) — это целая группа наследственных, генетических заболеваний, при которых страдают двигательные нейроны передних рогов спинного мозга. Из-за мутации в гене SMN1 у больных СМА снижена выработка SMN-белка, без которого нейроны разрушаются и перестают передавать сигналы от мозга к мускулатуре. В результате у человека постепенно атрофируются мышцы конечностей, головы и шеи.
Какой тип СМА самый опасный?
Существует 5 основных типов спинальных мышечных атрофий. Каждый из них имеет свои особенности, в том числе – симптомы и время манифестации.
Во многом тяжесть и скорость развития болезни определяется количеством у ребенка копий гена SMN2, он почти идентичен гену SMN1 и может в некоторой степени компенсировать его поломку.
Высокая смертность в случае отсутствия раннего лечения характерна только для СМА нулевого и первого типов. Большая часть пациентов с другими типами атрофий доживает до взрослого возраста. Четвертый тип, затрагивающий взрослых, на продолжительность жизни значительного влияния не оказывает.
Почему именно я? Как часто врачи ставят страшный диагноз
Обычно тип наследования СМА у детей — аутосомно-рецессивный. Некоторые, более редкие виды СМА, имеют другой тип наследования, а могут вообще не передаваться следующим поколениям. Это значит, что ребенок может заболеть только в том случае, если у обоих родителей есть поломанный ген, ответственный за синтез соответствующего белка. И даже тогда вероятность болезни составляет лишь 25%. Еще в четверти случаев ребенок вообще не унаследует мутаций, и в половине — сам станет бессимптомным носителем.
Согласно последним данным, в среднем дефектные гены, вызывающие мышечную атрофию, можно найти у каждого пятидесятого новорожденного, что не так мало, как хотелось бы. Сегодня среди 10 тысяч младенцев СМА диагностируют хотя бы у одного. Не зря эта болезнь является одной из ведущих причин детской смертности, связанных с генетическими нарушениями.

Когда стоит заподозрить неладное? Симптомы болезни
СМА первого типа может стать заметна почти сразу после рождения. Младенцы с этой тяжелой патологией:
Дети с СМА первого типа сначала могут выглядеть совершенно здоровыми. Однако со временем родители замечают, что ребенок мало двигается, быстро набирает вес, ему все труднее держать игрушки. Так как начинать лечить мышечную атрофию нужно как можно раньше, появление любых описанных симптомов — повод немедленно обратиться к врачу-неврологу.
Дети с СМА второго типа начинают отставать в развитии после 6–7 месяцев. Симптомы могут развиться еще позже, но обычно — до достижения ребенком 18 месяцев жизни. Дети с этой формой спинальной атрофии:
СМА третьего типа возникает к 1,5–2 годам, иногда — в позднем детстве или в подростковом возрасте. Ее основные признаки:
Взрослые, столкнувшиеся с СМА четвертого типа, как правило, жалуются на:
Эта форма СМА также способна прогрессировать, но почти никогда не затрагивает мускулатуру, отвечающую за дыхание, а потому не составляет угрозы жизни.
Может ли болезнь пройти сама по себе?
СМА — это расстройство, вызванное специфической мутацией в геноме. Оно всегда имеет прогрессирующий характер и не может пройти со временем, так как наша ДНК не способна починить сама себя, избавившись от нежелательных элементов кода.
Лечение СМА. Спинраза — это единственная надежда?
До недавнего времени вылечить СМА было невозможно, и все действия сводились к поддержанию жизни и улучшению ее качества. Для этого, например, используются специальные упражнения и приборы, призванные помочь двигаться и дышать, в том числе — зонды для кормления.
Настоящим прорывом стала разработка первого эффективного лекарства против спинальной мышечной атрофии. Препарат Спинраза (нусинерсен) был одобрен в США в 2016 году, а в России — в 2019-м. Он направлен на лечение всех типов СМА и подходит для пациентов всех возрастов. В основе — небольшие последовательности нуклеотидов, позволяющих синтезировать белок SMN на базе другого, схожего гена в нашей ДНК — SMN2. Препарат вводят в спинномозговую жидкость. Одной инъекции недостаточно. Чтобы защитить нейроны, лекарство нужно использовать регулярно: несколько раз в первый год жизни, и каждые 4 месяца — после.
В прошлом году в России стал доступен Рисдиплам. Он, как и Спинраза, воздействует на прочтение гена SMN2, но его можно пить в виде таблеток, что гораздо удобнее.
Третий препарат против СМА — Золгенсма (онасемноген абепарвовек), в нашей стране пока не зарегистрирован. Его применяют у детей до 2 лет. Внутри лекарства — аденовирусный вектор (оболочка вируса), который доставляет в нейроны работающий ген SMN1, после чего клетки начинают синтезировать нужный им белок (ДНК ребенка при этом никак не изменяется). Так как это средство генной терапии, для полного выздоровления хватает всего лишь одной дозы лекарства, в чем его значительное преимущество.
Сколько стоит лечение и когда его не поздно начать?
Ситуация для больных детей усугубляется в связи с тем, что лечить СМА нужно на ранних стадиях, так как из-за болезни нейроны отмирают постоянно, день за днем. Если ребенок болен СМА первого типа, у него есть в запасе около двух лет, у маленьких пациентов с другими формами болезни времени для лечения чуть больше, но прогноз тем лучше, чем раньше начата терапия. И все же на сегодняшний день прогресс в лечении СМА очевиден. Этот диагноз – больше не приговор, а в будущем спасительные лекарства, несомненно, будут становиться все эффективнее и дешевле.
Самое частое из редких: что нужно знать о СМА
С помощью фонда «Семьи СМА» мы собрали главные факты об этой болезни, которые нужно знать
Частота заболевания
Причина СМА
СМА – наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN – протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
 Изображение с сайта bfm.my
Изображение с сайта bfm.my
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена – SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения – не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА IV, этот тип называется ещё «взрослой СМА», поскольку болезнь проявляется обычно в возрасте после 35 лет.
Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.
Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук. Обычно проблем с глотательной и дыхательной функцией у больных нет.
Большинство из больных IV типом СМА могут ходить, и лишь некоторым приходится прибегать к инвалидным коляскам.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными – они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
В последние годы в мире активно разрабатываются препараты против СМА, три из них уже применяются в мире, остальные находятся на разных стадиях разработки.
Спинраза – препарат, разработанный компанией Biogen, увеличивает производство белка SMN из «резервного» гена SMN2. При регулярной терапии спинраза приостанавливает развитие СМА и стабилизирует состояние больного.
Спинраза выпускается в дозировке 12 мг на 5 мл для интратекального введения (непосредственно в спинномозговую жидкость) и одобрен для всех возрастов и типов СМА без каких-либо ограничений.
В первый год необходимо будет сделать 6 инъекций: 3 дозы с 14-ти дневным интервалом, 1 дозу спустя 30 дней и далее – раз в 4 месяца. Впоследствии препарат нужно будет применять постоянно в течение всей жизни каждые 4 месяца.
Препарат следует начать применять, как только был поставлен диагноз. Эффективность и безопасность применения нусинерсена изучалась у детей в возрасте от 0 до 17 лет, опыт применения у пациентов старше 18 лет ограничен, применение препарата у пациентов старше 65 лет не изучалось.
После применения спинразы наблюдались улучшения двигательной активности у пациентов со СМА I, II и III типа. Возможные осложнения спинразы – инфекция верхних дыхательных путей, инфекции нижних дыхательных путей и запоры, возможно также развитие ателектаза, отклонения коагуляции и тромбоцитопения, включая острую тяжелую тромбоцитопению, а также развитие гломерулонефрита.
13 февраля 2019 года спинраза получила в России орфанный статус. 16 августа 2019 года препарат был официально зарегистрирован в России.
3 августа 2020 комиссия Минздрава рекомендовала правительству включить препарат для лечения спинальной мышечной атрофии в перечень жизненно необходимых и важнейших лекарственных препаратов на следующий год. Однако по состоянию на сентябрь переговоры по возмещению за счет средств федерального бюджета на 2021 год не завершены.
Есть прецеденты выигранных судов, когда суд обязал региональные власти обеспечить пациентов спинразой за счёт региональных бюджетов.
Стоимость препарата – около 8 миллионов рублей за одну инъекцию.
Золгенсма (Zolgensma) — первый препарат генной терапии, разработанный для лечения спинальной мышечной атрофии (СМА). Предназначен для устранения генетической причины СМА путем замены дефектного или отсутствующего гена SMN1 для остановки прогрессирования заболевания. Препарат доставляет полностью функциональную копию гена SMN в организм человека. Препарат разработан компанией Авексис для однократного применения (одна инъекция на всю жизнь).
В настоящее время препарат одобрен в США для больных СМА возрастом до 2 лет, включая тех, кто не имеет симптомов при постановке диагноза. В Европе препарат получают больные СМА I типа с количеством копий SMN2 не более 3 копий, ограничения по весу 21 кг. Препарат вводится однократно внутривенно, доза определяется с учётом массы тела ребёнка.
Наиболее эффективно применение препарата до появления первых симптомов заболевания у детей, у которых наличие СМА установлено по результатам скрининга. Исследования о применении препарата у детей более позднего возраста, уже имеющих симптомы СМА, продолжаются.
Наиболее частыми нежелательными реакциями на фоне применения Золгенсма были повышение активности печёночных ферментов и рвота. В инструкции по применению препарата имеется предупреждение о риске развития тяжёлого острого поражения печени.
В середине июля 2020 года компания «Новартис» подала в Министерство здравоохранения РФ досье на регистрацию препарата к применению в России. По существующей процедуре, процесс регистрации может занять до полугода.
В настоящее время компанией-производителем объявлена программа сострадательного применения (до официальной регистрации препарата), по которому 100 доз препарата будут распространены между ста пациентами со СМА любого типа до двух лет методом лотереи. В России по этой программе препарат получили 4 ребёнка.
Кроме того, за счёт средств благотворителей препарат получили 16 российских детей, ещё двое принимают участие в программе клинических испытаний.
Стоимость дозы препарата составляет около 152 миллионов рублей (самый дорогой препарат в мире).
Рисдиплам (RG7916) («Эврисди») – препарат компании Roche, является модификатором сплайсинга (генетической модификации) гена SMN2, увеличивающим экспрессию полноразмерных функциональных белков. В отличие от других препаратов, применяется перорально.
Исследования проводились с участием пациентов от 0 до 60 лет со СМА II и III типов. Улучшает моторную функцию пациентов. Противопоказаний на сегодняшний день не зарегистрировано.
18 марта 2020 года компания «Рош» подала заявку на рассмотрение для дальнейшей регистрации препарата в Министерство здравоохранения РФ.
Кроме того, в мире идут клинические испытания ещё нескольких препаратов – бранаплам, релдесемтив, SRK-015 и других.
По мнению врачей комбинация препаратов при лечении СМА возможна, но её целесообразность должна рассматриваться в каждом случае индивидуально.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.
Фото с сайта f-sma.ru Благотворительный фонд «Семьи СМА» помогает детям и взрослым со спинальной мышечной атрофией и другими нервно-мышечными заболеваниями и их семьям.
Фонд работает по всей территории России. Работа фонда имеет два основных направления – оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования – например, СМА 300.
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно. В этом отношении СМА – такая же «отложенная» болезнь как, например, миодистрофия Дюшенна или синдром Ретта, когда ребёнок, некоторое время развивавшийся в соответствии с нормой, теряет приобретённые ранее навыки и становится инвалидом.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
Сколько больных СМА в России?
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Кто в России помогает людям со СМА и их семьям
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Известные люди со СМА
Итальянка Симона Спиноглио родилась с наследственным заболеванием – спинальной мышечной атрофией 2 типа. Она с самого рождения не может ходить и передвигается только с помощью электрической коляски. Но ее жизнь полна и насыщенна; ничто не может помешать ее стремлению жить.
Симона работает на «горячей линии» итальянской Ассоциации «Семьи СМА» (Famiglies of SMA) и помогает детям и взрослым со СМА и другими нервно-мышечными заболеваниями.
Также Симона записала несколько популярных в итальянском сообществе СМА песен – о свободе делать то, что ты хочешь, несмотря на болезнь.
Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
В 2008 году собрала собственную музыкальную группу (распалась в 2010). В 2013 году приняла участие в конкурсе «Фактор А» на телеканале «Россия». Заняла второе место и получила персональную премию Аллы Пугачёвой «Золотая звезда Аллы». В 2017 году из-за недопуска России в конкурсную программу не смогла принять участие в конкурсе «Евровидение». Передвигается на коляске.
Программист из Владимира Валерий Спиридонов. Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).
Сегодня Валерий – член городской общественной палаты Владимира, эксперт по вопросам доступной среды, а также создатель собственного сообщества «Desire for life», рассказывающего о создании доступной среды и перспективных медицинских проектах. Валерий – участник многих телепрограмм на российском и зарубежном ТВ.
Типы СМА
SMN-связанная СМА, или 5qSMA, или проксимальная СМА обычно подразделяется на три категории. Условно можно выделить еще СМА0 (ноль) и СМА4. Таким образом, есть несколько основных типов СМА, и все они протекают по-разному. Процесс может развиваться в различные периоды жизни, иметь свои клинические особенности, свой характер течения, прогноз и необходимый объем помощи и поддержки.
Тип 1 – наиболее тяжелый, с самым ранним дебютом, тип 3 – наименее тяжелый, с поздним возрастом начала. Некоторые специалисты выделяют еще тип 4 для обозначения умеренной или мягкой СМА с дебютом во взрослом возрасте.
Специфика заболевания в том, что у каждого ребенка, даже в пределах одной группы, СМА протекает по-разному, индивидуально. Внешне это проявляется в возможном объеме движений – некоторые дети способны удерживать голову, немного поднимать руки и приподнимать ноги, а другие просто лежат в классической позе «лягушка» и могут только чуть-чуть шевелить стопами и пальчиками кистей рук.
Виктор Дубовиц, один из корифеев в области СМА-ведения, в 90-е годы предложил помимо обычной классификации использовать более усложненную шкалу. Например, есть СМА1 и ее подтипы будут 1.1, 1.2, 1.3, т.е. от 1.1 до 1.9. Такая схема используется в Италии.

Американская система основывается на шкале ABC, когда В – это классика, А – слабый тип, а С, соответственно, более сильный. Система с подтипами ABC наглядно описывает клинику СМА и позволяет подбирать для ребенка поддерживающую терапию в зависимости от подгруппы и соответствующих прогнозов. Этой системой начинают использоваться в России и других странах.
Важно! С помощью генетического теста тип СМА не определяется. Тип устанавливается на основе функциональных возможностей ребенка.
Симптоматика заболевания проявляется внутриутробно в отсутствии у плода двигательной активности. С рождения у ребенка выражена генерализованная мышечная гипотония с характерной позой «лягушки», резко снижена спонтанная двигательная активность. Сухожильные рефлексы не вызываются.
Как правило, эти дети долго наблюдаются педиатрами и неврологами с диагнозом перинатальная энцефалопатия. Иногда врачи связывают симптомокомплекс вялого ребенка с тяжелыми родами. Но все дети с перинатальной энцефалопатией и с последствиями тяжелых родов достаточно быстро и хорошо адаптируются, постепенно улучшаются, в отличие от детей со СМА.
Прогноз крайне неблагоприятный – дети погибают, как правило, в очень раннем возрасте (до шести месяцев) от интеркуррентных заболеваний (осложняющих течение основной болезни).
Обычно СМА0 и СМА1 объединяют.
СМА1
При I типе спинальной мышечной атрофии (тип Верднига-Гофмана) мать уже во время беременности может обратить внимание на позднее и слабое шевеление плода. С рождения у ребенка выражено распространенное снижение мышечного тонуса (синдром «вялого ребенка»). С первых месяцев жизни появляются слабость и атрофия мышц верхних и нижних конечностей, с последующим вовлечением мышц туловища и шеи. Такие изменения мышц приводят к тому, что дети не могут сидеть. Мышечные атрофии и подергивания мышечных волокон обычно маскируются хорошо выраженной подкожно-жировой клетчаткой. Характерным симптомом является мелкое дрожание (тремор) пальцев вытянутых ручек. Иногда обнаруживаются подергивания мышц языка.
Типичным признаком также является ослабление или полное исчезновение сухожильных рефлексов (коленный, ахиллов), ограничение нормальной подвижности в суставе, деформации скелета. Вследствие слабости межреберных мышц грудная клетка ребенка становится уплощенной. Так как в результате слабости мышц не происходит достаточной вентиляции легких, присоединяются частые респираторные инфекции, возникают разнообразные дыхательные расстройства. Психическое развитие детей не страдает.
У младенцев могут возникать дыхательные нарушения и невозможность приема пищи. Врожденные контрактуры, варьирующие по степени выраженности от простой косолапости до генерализованного артрогрипоза (множественная врожденная патология аппарата движения, проявляющаяся многочисленными контрактурами суставов, мышечной гипотрофией и поражением спинного мозга), возникают примерно у 10 % новорожденных с тяжелым поражением. Дети грудного возраста лежат в расслабленной позе «лягушка», спонтанная двигательная активность снижена, дети не могут преодолеть силу тяжести конечностей, плохо удерживают голову.
Обычно, за исключением очень редких, тяжелых случаев, диагноз СМА не ставится в роддоме. Ребенка выписывают домой здоровым, и когда родители замечают низкий мышечный тонус, они или не придают этому серьезного значения, если не знают, как должен двигаться здоровый малыш, или успокаиваются советами доктора в поликлинике: «Не переживайте, все развиваются в свой срок, еще встанет и побежит». Родители могут не понимать всей тяжести проблем, это должен увидеть врач, но, к сожалению, в поликлиниках симптоматику СМА знают плохо.
Эти дети проявляют первые признаки заболевания в возрасте до 6 месяцев. Это значит, что они не приобретают навыки самостоятельно сидеть, ползать, ходить. В итоге, объем движений у таких детей очень мал. При этом СМА не затрагивает когнитивную сферу, дети все понимают, не затронута и чувствительность. Если с ними заниматься, как с обычными детьми – играть, читать, собирать пирамидки, – то эти дети в умственном и психическом плане развиваются абсолютно нормально, и все задержки развития, которые могут им ставить неврологи, связаны с нарушением двигательной активности и условиями, в которых они находятся.
СМА2
При спинальной мышечной атрофии II типа заболевание впервые проявляется несколько позже (в первые 1,5 года жизни ребенка) и характеризуется более медленным течением. Главным признаком является невозможность ребенка встать на ноги.
Дети со СМА2 обычно способны сосать и глотать, дыхательная функция в раннем младенческом возрасте не нарушена. Носовой оттенок голоса и нарушение глотания появляются в более старшем возрасте. Несмотря на прогрессирующую мышечную слабость, многие из них доживают до школьного и более старшего возраста, хотя на поздних стадиях заболевания отмечается тяжелая степень инвалидизации, и дети нуждаются в инвалидных колясках. Обычные коляски им не подходят, необходимы дополнительные поддержки, упоры, специальные приспособления, оптимально фиксирующие положение тела.
У многих пациентов с большой продолжительностью жизни одними из основных осложнений заболевания становятся сколиоз и контрактуры, которые развиваются очень быстро. Даже у лежачих детей сколиоз развивается достаточно существенно, искривление позвоночника происходит не от нагрузки, а от слабости.
Дети со СМА2 некоторый период времени бывают достаточно стабильны, и родители могут поддерживать тот набор навыков, который этими детьми уже приобретен.
Есть такой термин «плато заболевания», который обозначает некий период, в течение которого дети достаточно стабильны, и значительного ухудшения состояния, прогрессирования болезни не происходит. Это может выглядеть так: дети набирают навыки, как все обычные дети, при «запуске» болезни перестают развиваться, а потом регресс состояния у них может идти очень медленно или, наоборот, очень быстро, у каждого ребенка по-своему. Или так: дети определенное время набирают навыки, потом происходит какое-то событие или болезнь, часть навыков теряется, и дальше наступает довольно долгое «плато», в течение которого могут быть даже незначительные улучшения, но потом наступают неизбежные ухудшения. Скорость прогрессирования болезни, продолжительность «плато» (или его отсутствие) и последующих ухудшений – все это очень индивидуально.
СМА3
Болезнь Кугельберга-Веландер – наиболее легкая форма спинальной мышечной атрофии (СМА III типа). Начинается она в возрасте от 1,5 до 17 лет. С такой болезнью люди живут дольше, прогрессия идет медленно. Атрофия мышц начинается с ног и далее распространяется на руки. В младенческом возрасте клинические проявления заболевания могут отсутствовать. Прогрессирующая слабость развивается в проксимальных отделах конечностей, особенно в мышцах плечевого пояса. Пациенты сохраняют способность к самостоятельной ходьбе. Симптомы слабости мышц бульбарной группы появляются редко. Примерно у 25 % пациентов с этой формой СМА в большей степени выражена мышечная гипертрофия, а не атрофия мышц; поэтому может быть ошибочно диагностирована мышечная дистрофия. Пациенты могут дожить до зрелого возраста.
СМА у этой большой группы детей выявляется в возрасте старше полутора лет, т.е. обычно ребенок может самостоятельно ходить. Болезнь проявляется настолько индивидуально, что диагноз могут поставить в полтора года, а могут и в девять лет. В зависимости от этого будут разные прогнозы и по продолжительности, и по качеству жизни.
У детей со СМА3 продолжительность жизни почти не отличается от стандартной, они доживают и до 30, и до 40 лет. У них не так быстро прогрессируют все симптомы, но с этими детьми тоже надо проводить и реабилитационные занятия, и занятий по физической терапии.
Сейчас есть достаточно большое количество взрослых пациентов со СМА, и это не четвертый тип, это дети со СМА2 и СМА3, которые выросли. У них возникают большие проблемы, которые не связаны с перемещением и дыханием. Сколько бы они ни жили, 20 лет или 30, они уже разочаровались в медицине, потому что никто не знает, что с ними делать. Каждая мама ребенка со СМА и каждый взрослый пациент со СМА может рассказать примерно одну и ту же историю про прием у доктора: «Не приходите ко мне, мы не знаем, что с вами делать»; «Ты еще не умер, ах, ничего себе». Действительно, врачи не знают, что делать с этими пациентами, и говорят: «Ну, не лечится же, что ты хочешь от меня, правда, по-честному, я не знаю, что с тобой делать». Ими никто не занимается, после 18 лет вообще не знают, что с ними делать.
Эти пациенты (часто невидимые пациенты, так как сидят дома) не верят в медицинскую помощь, потому что всю жизнь от них отмахивались. И они обращаются за помощью только тогда, когда уже невозможно игнорировать симптоматику, когда сильная боль, т.е. поздно – практически уже на смертном одре. До недавнего времени с этой категорией пациентов не было никакой работы. Сейчас появилась возможность как-то улучшать ситуацию, больше помощи оказывают благотворительные фонды, этой проблеме уделяется больше внимания.
Заболевание СМА3 развивается не очень стремительно, постепенно происходит общее ослабление организма и медленная потеря приобретенных навыков.
СМА4
Четвертый тип СМА проявляется у людей старше 25 лет. Заболевание развивается довольно медленно, практически не влияя на продолжительность жизни. При СМА4 может развиваться тремор, возникать общее ослабление, в том числе, и мышечной силы. Со временем СМА4 может приводить к потере способности к самостоятельному передвижению.


