Болезнь Баттена: ранние симптомы и диагностика
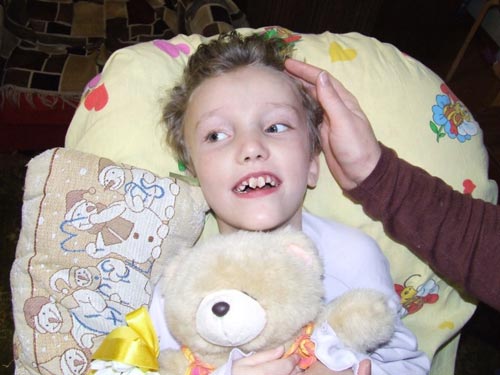
Болезнь Баттена — это наследственное неврологическое заболевание, которое часто приводит к летальному исходу. Как правило, оно развивается еще в детстве, в возрасте от 5 до 10 лет. Ранние симптомы включают судороги, ухудшение зрения, незначительные изменения в поведении пациента.

Патология и проявления болезни Баттена
Болезнь Баттена является наиболее распространенным заболеванием, известной под общим названием «Нейрональные цероидные липофусцинозы» (НЦЛ). Первоначально болезнь Баттена использовалась для описания ювенильной формы НЦЛ, но теперь чаще используется для обозначения всех форм НЦЛ.
Симптомы болезни Баттена вызваны скоплением в тканях организма веществ, называемых липопигментами, составными частями жира и белка.
Эти липопигментные отложения накапливаются:
В конце концов у детей развиваются умственные нарушения, судороги, ухудшается зрение и моторные навыки, пока они не станут слепыми и практически полностью обездвиженными.
Эпидемиология и наследственность при болезни Баттена
Данное заболевание относится к категории редких недугов. Болезнь Баттена наследуется по аутосомно-рецессивному типу.
Это означает, что ребенок должен унаследовать два дефектных гена (по одному от каждого родителя) для проявления и развития этой болезни.
Когда оба родителя являются носителями дефектного гена, у каждого ребенка этой пары есть шанс унаследовать заболевание.
Ткань исследуется с помощью электронного микроскопа для выявления отложений НЦЛ. Могут использоваться образцы:
Нейрональный цероидный липофусциноз
Нейрональный цероидный липофусциноз (НЦЛ) – это группа генетических заболеваний, в основе которых лежит накопление в клеточных структурах нейронов и других тканей токсического пигмента – липофусцина. Патология наследуется по аутосомно-рецессивному типу.
Патогенез нейронального цероидного липофусциноза
В основе патогенеза лежит нарушение утилизации пигмента липофусцина. Он накапливается в тканях организма человека и в норме, но гораздо медленнее. В случае нейронального цероидного липофусциноза это накопление происходит стремительно и приводит к атрофии тканей. Липопигменты локализуются в клеточных органеллах – лизосомах, выполняющих функцию утилизации отработанных клеточных элементов.
Ранее в основе классификации нейронального цероидного липофусциноза лежал возраст пациента, в котором начинало манифестировать заболевание. В зависимости от этого выделяли 4 типа патологии:
По мере изучения наследственных болезней были выявлены мутации в генах NCL, которые и легли в основу современной классификации. В настоящее время выделяют 10 типов заболевания в зависимости от имеющихся генетических нарушений.
Клинические проявления нейронального цероидного липофусциноза
Нейрональный цероидный липофусциноз является прогрессирующим заболеванием детей и подростков, однако некоторые формы патологии могут развиваться и у взрослых людей младше 40 лет. До появления клинических проявлений дети ничем не отличаются от своих сверстников.
Манифестация заболевания проявляется одним из трех признаков:
По мере прогрессирования присоединяются следующие признаки:
Со времени возникновения первых симптомов состояние ребенка быстро ухудшается. Гибель происходит в течение нескольких лет. При благоприятных формах нейронального цероидного липофусциноза пациенты редко переживают сорокалетний рубеж.
Методы лечения нейронального цероидного липофусциноза
На сегодняшний день эффективного патогенетического лечения нет, однако состояние пациентов можно облегчить с помощью симптоматической терапии. Это назначение противоэпилептических препаратов, миорелаксантов, седативных, антипсихотических, обезболивающих средств, витаминов. Иногда временное улучшение приносит физиотерапия.
В некоторых странах ведется активный поиск методов лечения, который сосредоточен на трех направлениях:
В целом есть обнадеживающие результаты, позволяющие надеяться на победу страшной болезни в будущем. В США и Европе зарегистрирована фермент-заместительная терапия рекомбинантной альфа-церлипеназой (Бринейра). Препарат позволяет прекратить прогрессирование заболевания.
В рамках диагностического поиска используется нейровизуализация (МРТ, ПЭТ), ЭЭГ, электроретинограмма. Активно применяется гистологическое исследование, но наиболее ценной информацией обладает молекулярно-генетическая диагностика для определения мутаций в генах. Исследуется материал от обоих родителей и ребенка. На основании полученных данных делается заключение о наличии или отсутствии у пациента нейронального цероидного липофусциноза.
Государство есть не везде
Дети с болезнью Баттена выживут только в Москве и Чечне?


32 человека – столько сейчас живет в России детей с диагностированным орфанным заболеванием «нейрональный цероидный липофусциноз 2-го типа» (НЦЛ-2), известным также как болезнь Баттена. Болезнь тяжелая: без необходимого лечения пациенты не доживают до взрослого возраста. Москва и Чеченская Республика сегодня единственные регионы, где родителям не нужно добиваться жизненно важного лекарства в судах. В других субъектах РФ на суды уходит время, во многих случаях состояние детей непоправимо ухудшается.
Болезнь Баттена входит в перечень орфанных заболеваний Минздрава РФ как «болезнь эндокринной системы, расстройства питания и нарушения обмена веществ». Особенности течения: проявляется не раньше 23 лет, поражает клетки головного мозга. Из-за этого у пациентов начинаются эпилептические приступы, ухудшаются интеллект, двигательные функции, речь, зрение. К 78 годам дети с болезнью Баттена не встают с постели, умирают в 1012 лет.
Единственный препарат, способный остановить развитие заболевания, – церлипоназа альфа, появившийся на медицинском рынке в 2017 году под торговым названием бринейра. В России лекарство не зарегистрировано (в данный момент проходит процедуру регистрации) и стоит дорого: годовой курс лечения обходится около 5060 млн руб. Причем принимать препарат нужно пожизненно, один раз в две недели. Лекарство вводится прямо в головной мозг пациента с помощью специального порта, установка которого требует хирургического вмешательства.
Церлипоназа альфа не возвращает утраченные функции организма – препарат стабилизирует текущее состояние, продлевает жизнь и повышает ее качество. Чем раньше начинается терапия, тем лучше.
То лучше, то хуже
У Варвары Муфахаровой из города Гулькевичи Краснодарского края болезнь проявилась в январе 2019-го, когда ей было три года. Эпилептические приступы случались так часто, что у девочки, прежде не отстававшей в развитии от сверстников, отказали ноги, она почти перестала говорить.
Точный диагноз – НЦЛ-2 – семья узнала только спустя полгода, после сдачи генетического анализа. Почти сразу врачи назначили препарат церлипоназа альфа. Однако Минздрав Краснодарского края не обеспечивает лекарством маленькую пациентку, несмотря на решение суда «к немедленному исполнению».
Суд состоялся в январе 2020 года. В конце марта Министерство здравоохранения Краснодарского края получило разрешение на ввоз необходимого Варваре лекарства, но вместо объявления государственных торгов на закупку обратилось к производителю церлипоназы альфа с просьбой предоставить препарат бесплатно.
Фармацевтическая компания BioMarin, которая производит церлипоназу альфа, выделила несколько флаконов лекарства, но при одном условии: лечение Варвары Муфахаровой безо всякого перерыва обязательно подхватит краевой минздрав.
В начале июля девочка получила первую инфузию препарата. После второго приема, по словам мамы Татьяны Митейко, Варя начала ползать. После третьего и четвертого вливания стала снова учиться ходить. Заново вспоминала слова, которые перестала произносить. Когда лекарства оставалось еще на два вливания, в минздраве родителям сообщили: средства на закупку препарата ищут. Последняя инфузия прошла в конце сентября. Следующая была запланирована через две недели, но лекарство минздрав не предоставил.
В конце октября Минздрав Краснодарского края в очередной раз ответил родителям Варвары Муфахаровой: «Учитывая. что лечение необходимо проводить дорогостоящим препаратом. изыскиваются финансовые средства для проведения процедуры закупки».
Состояние девочки с момента последней инфузии ухудшилось. Варвара Муфахарова почти перестала есть и спать. Ее опять не держат ноги. Вернулись частые эпилептические приступы.
– Церлипоназа альфа – препарат серьезный, его нельзя сначала назначить, сделать несколько инфузий, а потом без медицинского заключения прекратить лечение, – говорит Анастасия Татарникова, руководитель автономной некоммерческой организации «Дом редких», которая помогает российским пациентам с орфанными заболевания, в том числе детям с НЦЛ-2, защищать свои права. – Кроме того, врачи сразу заметили улучшение состояния Вари. Это не рядовая ситуация, потому что эффект церлипоназы альфа обычно оценивают не ранее чем через полгода после начала лечения.
Лечащие врачи девочки еще в сентябре сообщили в Минздрав Краснодарского края о том, что их маленькой пациентке значительно лучше.
– Даже в этом смысле у ведомства не может быть сомнений в необходимости препарата, – подчеркивает Анастасия Татарникова.

В неизвестности
Насколько долго Варе Муфахаровой придется ждать лекарства и насколько необратимы будут ухудшения в ее случае – неизвестно. Ее маме в Министерстве здравоохранения Краснодарского края обещают, что деньги вот-вот выделят или уже выделили, но подтверждения этому пока нет. Семья надеется на лучшее: в соседнем же регионе, в Ростовской области, лекарство для пятилетнего мальчика с НЦЛ-2 закупили на средства госбюджета. И гораздо быстрее, чем Варе Муфахаровой: суд прошел в марте 2020 года, позже, чем в Краснодарском крае, а пациент лечится с октября.
Правда, есть и другой пример, когда с препаратом региональный минздрав откровенно опоздал.
В Екатеринбурге семья Файдаровых два с половиной года – с середины 2017-го по конец 2019-го – добивалась покупки церлипоназы альфа для своих двоих детей-погодок – Дианы и Демида. У Дианы болезнь была выявлена раньше из-за начавшихся ухудшений. У Демида тяжелых проявлений заболевания еще не было. Чтобы его спасти, зафиксировать состояние, нужно было вводить назначенный врачами дорогой препарат. Сейчас дети Файдаровых получают лекарство, но Диана питается через зонд, а Демид уже не поднимается с постели.
В настоящий момент 12 детей из разных регионов России получают церлипоназу альфа. Пенза, Екатеринбург, Новосибирская, Владимирская, Воронежская, Ростовская и Московская области, Башкортостан, Хакасия – во всех этих регионах родителям пришлось судиться за лекарство и долго обивать пороги местных минздравов, чтобы их дети начали получать препарат бесплатно.
– Часто местные минздравы отказываются закупать лекарство из-за того, что оно не зарегистрировано в России, – объясняет юрист АНО «Дом редких» Кирилл Куляев. – Однако это не может становится причиной отказа, заявил Верховный суд в сентябре 2019 года во время процесса, где решалось, обеспечивать ли препаратом девочку с НЦЛ-2. Родители дошли до высшей инстанции, чтобы доказать свою правоту.
Решение Верховного суда сейчас помогает выигрывать в судах первой инстанции, однако региональные минздравы чаще всего подают апелляции, ждут решения суда второй инстанции, чтобы начать закупать препарат. Единственное исключение – Новосибирск: здесь церлипоназу альфа для ребенка купили после первого же судебного решения.
От чего зависит, насколько быстро будет исполняться решение суда? Юрист полагает, от политической воли в конкретных области или городе, человеческого фактора. Степень обеспеченности региона или его дотационность не всегда влияют на скорость финансовых поисков в бюджете, объявления закупок для обеспечения детей препаратом.
– Например, в Башкортостане были серьезные трудности с обеспечением ребенка непрерывной терапией, – рассказывает Кирилл Куляев. – Семье пытались помочь в прокуратуре, вмешался Следственный комитет, главу местного минздрава три раза привлекали к административной ответственности. Препарат в конце концов начали закупать, но по одному флакону, в лечении мальчика были перебои. Сейчас обеспечение наладилось, но что будет в следующем году, сказать трудно.
В Воронежской области, в городе Семилуки, тоже живет ребенок с НЦЛ-2. Его мать долго добивалась церлипоназы альфа для сына: местные СМИ описывали почти детективную историю, как врачи сначала согласились с тем, что препарат показан мальчику, а затем отказались от своего экспертного решения.
Почти в таком же положении, как Варя Муфахарова, сейчас находится пятилетняя Василиса Седова из Оренбурга. Единственное отличие: девочке еще не начинали вливать лекарство. В апреле мама Василисы, Анна Седова, выиграла суд, который встал на сторону маленькой пациентки с редким заболеванием. В сентябре апелляция оставила решение в силе. Пока региональный минздрав не торопится исполнять его, Василисе становится хуже.
Ни один родитель не откажется от борьбы за жизнь собственного ребенка. Но когда региональное здравоохранение сдастся, может быть уже поздно.
Куда переезжать?
Первая инфузия препарата церлипоназа альфа в России состоялась 26 августа 2019 года. Лекарство получила трехлетняя Лера Змейкова. Вливание прошло в одной из московских больниц, препарат девочке выдали по назначению врачей.
Лера, получившая лекарство в Москве, родом из Екатеринбурга – чтобы добиться помощи для дочери, пока заболевание не успело разрушительно отразиться на ее организме, родители практически сразу после постановки диагноза переехали в столицу. Они видели, к чему может привести долгое выбивание препарата из свердловского минздрава на примере брата и сестры Файдаровых и не хотели подобного повторения истории для Леры.
Лера Змейкова получает препарат регулярно, без перерывов и не отличается от остальных детей. В ее жизни существует только одно небольшое неудобство: приходится раз в две недели на три дня ложиться в больницу для проведения инфузии.
В чеченском селе Бено-Юрт девочка с НЦЛ-2 получила препарат после переписки родителей с республиканским минздравом, тоже без вмешательства суда.
В Москве кроме Леры Змейковой есть еще один ребенок с НЦЛ-2, его семья тоже переехала сюда из другого региона. Но переезжать в столицу всем семьям, где есть «редкие» дети, не выход, считает Кирилл Куляев:
– Если все нуждающиеся будут ехать за спасением в другой регион, пузырь орфанного обеспечения там очень быстро лопнет. Переезжать всем – это абсурд.
Известны случаи, когда российские семьи еще более кардинально меняли регион проживания ради лечения ребенка.
Анастасия Татарникова рассказывает о маме двоих сыновей с НЦЛ-2, которая переехала с детьми в Германию:
– Ее младшего сына позвали на клинические испытания церлипоназы альфа для детей до двух лет – старший не подходил по возрасту, но женщина все равно решила уезжать. Ей пришлось выбирать жизнь хотя бы для одного ребенка. Мы на связи с семьей, мама говорит, что очень скучает по родине, но высоко отзывается об организации медицинской помощи в Германии, в том числе паллиативной, которая уже необходима ее старшему сыну.
Дети из еще двух российских семей были приглашены на клинические исследования. После их завершения прием препарата церлипоназа альфа должен продолжаться, поэтому семьи решили обосноваться в Европе. Родители рассудили, что за границей шансов у их детей выжить гораздо больше.
Для того чтобы изменить ситуацию к лучшему и не заставлять семьи с тяжелобольными детьми ждать или переезжать в другую страну или в другой регион, необходимо переводить лекарственное обеспечение орфанных заболеваний, в том числе НЦЛ-2, на федеральный уровень, считают сотрудники АНО «Дом редких».
Сейчас в России создается фонд помощи детям с редкими болезнями, в октябре Анастасия Татарникова принимала участие в общественном обсуждении его возможной работы. Она надеется, что НЦЛ-2 войдет в приоритетный для финансирования список заболеваний, который будет сформирован Минздравом РФ.
Нцл что за болезнь
НЕЙРОНАЛЬНЫЙ ЦЕРОИДНЫЙ ЛИПОФУСЦИНОЗ 2 ТИП.
(НЦЛ 2, НЕДОСТАТОЧНОСТЬ ТРИПЕПТИДИЛ ПЕПТИДАЗЫ 1, КЛАССИЧЕСКИЙ ВАРИАНТ ПОЗДНЕЙ МЛАДЕНЧЕСКОЙ ФОРМЫ НЦЛ 2 ТИПА, БОЛЕЗНЬ ЯНСКОГО-БИЛЬШОВСКОГО, сLINCL).
CLN 2; CEROID LIPOFUSCINOSIS, NEURONAL, 2; CLN2; CEROID LIPOFUSCINOSIS, NEURONAL, 2, VARIABLE AGE AT ONSETJAN SKY-BIELSCHOWSKY DISEASE; NEURONAL CEROID LIPOFUSCINOSIS, LATE INFANTILE, INCLUDED; LINCL, INCLUDED
Генетика : мутации гена трипептидил пептидазы 1 (TPP1 MIM *607998). Ген картирован на коротком плече 11 хромосомы (локус 11p15).
Эпидемиология :суммарная частота встречаемости всех форм НЦЛ в мире составляет 1: 25000.
Тип наследования :аутосомно-рецессивный
Патогенез :при всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента, который состоит из белков сапозинов А и D и/или субединицы с митохондриальной АТФ синтазы. Накапливаемый материал характеризуется аутофлюоресценцией в зелено-желтом спектре при возбуждении светом длиной от 340 до 360 нм. При гистохимическом окрашивании материал дает положительную реакцию на кислую фосфатазу и окрашивается суданом черным В, что свидетельствует о присутствии фосфолипидов. Он также является ШИК- положительным, что может указывать на высокое содержание углеводов. Большинство из этих свойств идентично свойствам так называемых пигментов «изнашивания», или «старения», обычно называемых липофусцинами или цероидами. При электронной микроскопии нейрональных и экстранейрональных тканей обнаруживают цитосомы, заполненные агрегатами характерной морфологии, описываемые как «криволинейные» профили, по типу «отпечатков пальцев», «прямолинейные» профили, гранулярные осмиофильные отложения.
Клинические проявления :в зависимости от возраста манифестации НЦЛ 2 типа подразделяют на три формы: врожденную, позднюю младенческую, юношескую формы. Классическая форма поздней младенческой формы НЦЛ широко распространена во всем мире, но с наибольшей частотой в западной Финляндии. Обычно первые симптомы появляются в возрасте от двух до четырех лет. Манифестными симптомами являются генерализованные тонико-клонические приступы, задержка речевого развития, атаксия. По мере развития заболевания присоединяются другие типы эпилептических приступов (миоклонические, парциальные, диалептические). Вслед за развитием эпилептических приступов развиваются интеллектуальные нарушения, и происходит утрата ранее приобретенных двигательных навыков. Часто у пациентов наблюдаются атаксия, мышечная гипотония, дизартрия. Зрительные нарушения, как правило, присоединяются позднее и прогрессируют очень медленно. Мышечная гипотония сменяется спастичностью, формируются сгибательные контрактуры конечностей, формируется псевдобульбарный синдром. В терминальную стадию болезни те же симптомы, что и при младенческой
Диагностика :основными методами подтверждения диагноза НЦЛ 2 типа являются определение активности ферментов в лейкоцитах крови или культуре клеток кожных фибробластов, молекулярно-генетические наследования, исследования биоптатов при электронной микроскопии
Нцл что за болезнь
НЕЙРОНАЛЬНЫЙ ЦЕРОИДНЫЙ ЛИПОФУСЦИНОЗ 1 ТИП
(НЦЛ 1 ТИПА; НЕЙРОНАЛЬНЫЙ ЦЕРОИДНЫЙ ЛИПОФУСЦИНОЗ ИНФАНТИЛЬНАЯ ФОРМА; БОЛЕЗНЬ САНТУВУОРИ-ХАЛТИА, INCL, НЕДОСТАТОЧНОСТЬ ПАЛЬМИТОИЛ ТИОЭСТЕРАЗЫ)
CLN1; CEROID LIPOFUSCINOSIS, NEURONAL, 1; CLN1;CEROID LIPOFUSCINOSIS NEURONAL 1 VARIABLE AGE AT ONSET; NEURONAL CEROID LIPOFUSCINOSIS, INFANTILE, INCLUDED; INCL, INCLUDED; SANTAVUORI DISEASE, INCLUDED; SANTAVUORI-HALTIA DISEASE, INCLUDED
Генетика : мутации гена пальмитоил тиоэстеразы 1 (PPT1; MIM *600722).Ген картирован на коротком плече 1 хромосомы (локус 1p32).
Тип наследования :аутосомно-рецессивный
Эпидемиология :суммарная частота встречаемости всех форм НЦЛ в мире составляет 1: 25000.
Клинические проявления : в зависимости от возраста манифестации НЦЛ 1 типа подразделяют на три формы: врожденную, позднюю младенческую, юношескую и взрослую формы. При классической инфантильной форме НЦЛ первые симптомы заболевания обычно появляются в возрасте от 6 до 24 месяцев, в некоторых случаях, заболевание может дебютировать на первом полугодии жизни или после двух лет. Манифестными симптомами являются различные типы эпилептических приступов (генерализованные тонико-клонические, миоклонические), мышечная гипотония, задержка психо-моторного развития. С момента появления первых клинических симптомов достаточно быстро происходит регресс психо-моторного развития, судороги становятся резистентными к антиэпилептической терапии, развиваются мозжечковые расстройства, деменция и нарушается сон. К возрасту 24-36 месяцев наблюдается грубая деменция, декортикационная/децеребрационная ригидность, усиливаются миоклонии на различные внешние раздражители. Неблагоприятный исход через несколько лет от появления первых симптомов болезни.
Диагностика :при МРТ головного мозга у подавляющего большинства пациентов обнаруживают диффузную кортикальную и субкортикальную атрофию вещества головного мозга и мозжечка, а также повышение интенсивности МР сигнала в перивентрикулярном белом веществе и снижение интенсивности сигнала в области базальных ганглиев и таламусов. При инфантильной форме НЦЛ на КЭЭГ регистрируют уменьшение амплитуды основного коркового ритма, во время сна наблюдается отсутствие сформированных сонных веретен. Также обнаруживают ослабление реакции ЭЭГ на пробы с открыванием-закрыванием глаз с последующей депрессией альфа-ритма по сравнению с фоновой корковой активностью. Основными методами подтверждения диагноза НЦЛ1 типа являются определение активности ферментов в лейкоцитах крови или культуре клеток кожных фибробластов, молекулярно-генетические наследования, исследования биоптатов при электронной микроскопии. Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru).


